亲爱的科研小伙伴们,你们的小师妹又来啦!今天,我要给大家带来一个超级棒的生物信息学工具——dinoR包。在这个基因组学日新月异的时代,我们经常需要比较两组NOMe-seq实验中核小体结合/可及性模式的潜在差异。这不,小师妹最近就发现了这个超给力的R包,它不仅能够帮助我们高效地处理大量数据,还能让我们轻松地检测出不同样本间的细微差别。
想象一下,我们可以一次性对上千个感兴趣的基因组区域(ROIs)进行高覆盖率的NOMe-seq数据获取,这在以前可是想都不敢想的事情哦!而且,这个`dinoR`包还能让我们专注于比较具有相似特征的ROIs,比如转录因子结合位点,并对样本间的NOMe-seq模式差异进行统计分析。这简直就是我们这些科研小伙伴们的福音嘛!注意哦,这个R包操作占用内存比较大,建议使用服务器哦,欢迎联系小师妹租赁性价比高的服务器~
别急,小师妹这就带你一起探索`dinoR`的神奇功能,让你的研究工作更加得心应手。不管你是基因组学的老手,还是刚入门的新手,这个工具都能让你的研究之路更加顺畅。准备好了吗?让我们一起开启这场基因组探索之旅吧!如果你在操作过程中遇到任何问题,别犹豫,记得联系小师妹哦~
安装和加载R包
首先,让我们安装dinoR包并加载所有要用的包:
if (!require(“BiocManager”, quietly = TRUE)) {
install.packages(“BiocManager”)}
BiocManager::install(“dinoR”)
suppressPackageStartupMessages({
library(dinoR)
library(ggplot2)
library(dplyr)})
加载NOMe-seq数据
接下来,小师妹来给你简单说说怎么加载NOMe-seq数据吧!
首先,我们要加载的是ADNP敲除和野生型小鼠胚胎干细胞的NOMe-seq数据。这些数据是通过一系列复杂的步骤处理得到的,包括使用biscuit工具将读段映射到基因组,用UMI-tools去除重复的UMIs,以及用fetch-NOMe包获取甲基化保护信息。最后,我们用NOMeConverteR包把这些数据转换成了RangedSummarizedExperiment对象,这样就可以方便地在R中进行分析了。
在R中,我们可以通过以下简单的代码来加载这些数据:
data(NomeData)
# NomeData <- readRDS(file = “NomeData.rds”) #直接读取小师妹准备好的数据
NomeData
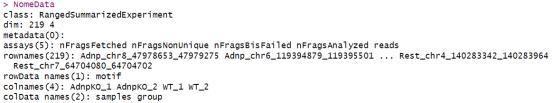
这个NomeData对象包含了219个感兴趣区域(ROIs)的信息,并且每个ROI都对应着4个样本的数据:两个ADNP敲除样本和两个野生型样本。这些ROIs都是围绕转录因子基序中心的,并且长度一致,这样我们就可以观察到甲基化保护的不对称性了。
每个样本的甲基化数据都存储在reads测定中,这是一个包含GPos对象的稀疏逻辑矩阵。这些数据告诉我们每个基因组位置上的甲基化保护情况,是被保护了还是被甲基化了。
接下来,我们来看看reads测定中包含的GPos对象,它在两个稀疏逻辑矩阵中包含了GCH甲基化数据,一个用于保护免受甲基化,另一个用于甲基化。
assays(NomeData)[[“reads”]][1, 1]
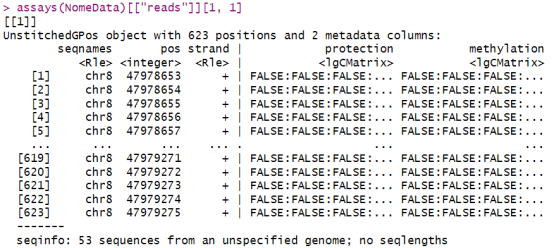
通过这些步骤,我们就能够加载并准备好NOMe-seq数据,为接下来的分析打下坚实的基础。如果你对这个过程有任何疑问,随时可以联系小师妹哦!
绘制meta plots
如果想要比较在REST、CTCF或ADNP这些转录因子结合位点中心的ROIs之间的差异,可以生成一种叫做“meta plots”的图,这种图可以帮助我们观察不同样本间在这些特定区域的甲基化保护模式。
我们可以使用来自野生型小鼠ES细胞的两个样本,以及来自ADNP敲除小鼠ES细胞的两个样本。如果某个ROI-样本组合的读数少于10个,我们会将其排除在外。
下面是生成这些图的R代码:
avePlotData <- metaPlots(NomeData = NomeData, nr = 10, ROIgroup = “motif”)
# 绘制平均图
ggplot(avePlotData, aes(x = position, y = protection)) +
geom_point(alpha = 0.5) +
geom_line(aes(x = position, y = loess), col = “darkblue”, lwd = 2) +
theme_classic() +
facet_grid(rows = vars(type), cols = vars(sample), scales = “free”) +
ylim(c(0, 100)) +
geom_hline(
yintercept = c(10, 20, 30, 40, 50, 60, 70, 80, 90),
alpha = 0.5, color = “grey”, linetype = “dashed”
)
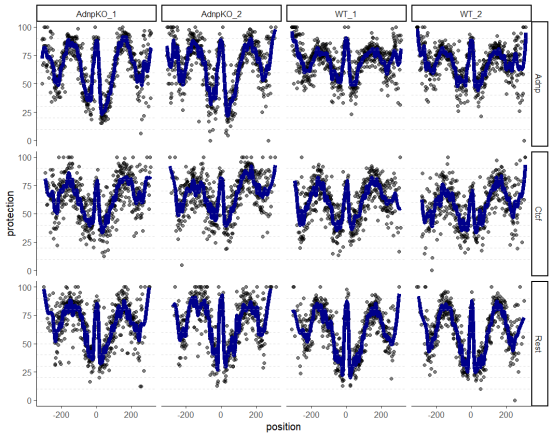
这段代码会生成一个包含所有样本和ROIs的图,其中每个点代表一个位置的保护水平,而蓝色的线则表示通过loess平滑处理后的趋势。我们还在图中添加了水平的虚线,以便于观察保护水平的变化。
从图中,我们可以观察到REST和CTCF结合位点周围的NOMe足迹没有变化,但在ADNP结合位点周围,野生型和ADNP敲除细胞之间存在明显的差异。这意味着ADNP的缺失可能会影响这些位点的核小体保护模式。
五种染色质模式的片段计数
接下来,我们来聊聊如何确定五种染色质模式的片段计数:转录因子结合位点(TF)、开放染色质、上游核小体(upNuc)、下游核小体(downNuc)和普通核小体(Nuc)。这些模式有助于我们理解基因组中不同区域的功能状态。
首先,我们采用了Sönmezer等人在2021年的研究方法,并稍作修改。我们根据围绕基序中心的三个窗口(-50:-25、-8:8、25:50)来分类每个片段,这些窗口对应于提供的ROIs的中心区域。
NomeData <- footprintCalc(NomeData)
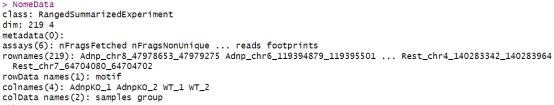
执行这个函数后,NomeData对象中会新增一个名为footprints的测定,这个测定会将每个片段归类为上述五种足迹模式之一。
assays(NomeData)[[“footprints”]][[1, 1]][1:10]
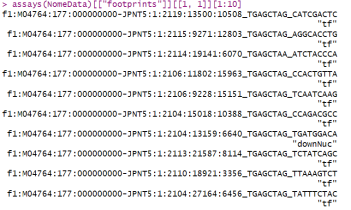
接下来,我们统计每个样本-ROI组合中支持每种足迹类别的片段数量。这会在NomeData中为每种足迹类别新增一个测定,并且还会新增一个包含所有足迹类别合并计数的测定(称为“all”)。
NomeData <- footprintQuant(NomeData)
assays(NomeData)[7:12]

请注意,如果一个片段在所有三个用于分类的窗口中都没有甲基化保护数据,那么这个片段将不会被使用。这样可以确保我们得到的数据是准确和可靠的。
最后,我们可以用这些数据来比较ADNP敲除和野生型样本之间足迹的丰度差异。这有助于我们理解ADNP基因的变化如何影响染色质的结构和功能。
比较ADNP敲除与野生型样本的NOMe-seq足迹丰度差异
接下来,我们要使用edgeR包来分析ADNP敲除(KO)和野生型(WT)样本之间每种足迹类型的片段计数与总片段计数的差异。我们会计算总片段计数的TMM(Trimmed Mean of M-values)标准化库大小。
首先,我们使用`diNOMeTest`函数来比较ADNP敲除和野生型样本之间的差异:
res <- diNOMeTest(NomeData,
WTsamples = c(“WT_1”, “WT_2”),
KOsamples = c(“AdnpKO_1”, “AdnpKO_2”))
这段代码会输出一个包含差异分析结果的数据框,显示了每种足迹类型在ADNP敲除和野生型样本之间的对数倍数变化(logFC)、对数标准化丰度(logCPM)、F统计量、P值、FDR(false discovery rate)以及对比组。
然后,我们可以简单地绘制每种ROI类型中受调控的ROI数量:
res %>%
group_by(contrasts, motif, regulated) %>%
summarize(n = n()) %>%
ggplot(aes(x = motif, y = n, fill = regulated)) +
geom_bar(stat = “identity”) +
facet_grid(~contrasts) +
theme_bw() +
theme(axis.text.x = element_text(angle = 90, vjust = 0.5, hjust = 1)) +
scale_fill_manual(values = c(“orange”, “grey”, “blue3”))

或者,我们可以用MA图来展示结果:
ggplot(res, aes(y = logFC, x = logCPM, col = regulated)) +
geom_point() +
facet_grid(~contrasts) +
theme_bw() +
scale_color_manual(values = c(“orange”, “grey”, “blue3”))
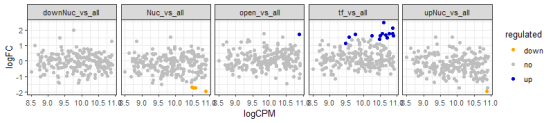
这些图表帮助我们直观地看到ADNP敲除和野生型样本之间在不同足迹类型上的丰度差异。通过这些分析,我们可以更清楚地了解ADNP基因对染色质足迹模式的影响。
探索ADNP敲除与野生型样本的染色质足迹差异
首先,我们要计算在ADNP敲除和野生型样本中,每种足迹类型的片段所占的百分比。这将帮助我们了解不同足迹类型在样本中的相对丰度。
footprint_percentages <- footprintPerc(NomeData)
fpPercHeatmap(footprint_percentages)
这段代码会生成一个热图,展示野生型和ADNP敲除样本中每种足迹类型的百分比。通过这个热图,我们可以直观地比较两组样本之间的差异。

接下来,我们要比较ADNP敲除和野生型样本之间足迹百分比的差异,并进行显著性测试。
compareFootprints(footprint_percentages, res,
WTsamples = c(“WT_1”, “WT_2”),
KOsamples = c(“AdnpKO_1”, “AdnpKO_2”), plotcols =
c(“#f03b20”, “#a8ddb5”, “#bdbdbd”))
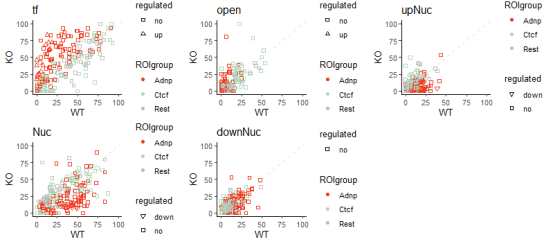
通过这个比较,我们可以发现在ADNP敲除样本中,围绕ADNP基序的转录因子足迹显著增加,而核小体足迹则减少。这表明ADNP基因的缺失可能会影响这些区域的染色质状态。
合并核小体模式
如果我们对上游和下游核小体模式不感兴趣,而是希望将所有核小体模式片段保留在核小体组内,我们可以通过设置`combineNucCounts = TRUE`来实现。
res <- diNOMeTest(NomeData,
WTsamples = c(“WT_1”, “WT_2”),
KOsamples = c(“AdnpKO_1”, “AdnpKO_2”), combineNucCounts = TRUE
)
footprint_percentages <- footprintPerc(NomeData, combineNucCounts = TRUE)
compareFootprints(footprint_percentages, res,
WTsamples = c(“WT_1”, “WT_2”),
KOsamples = c(“AdnpKO_1”, “AdnpKO_2”), plotcols =
c(“#f03b20”, “#a8ddb5”, “#bdbdbd”))
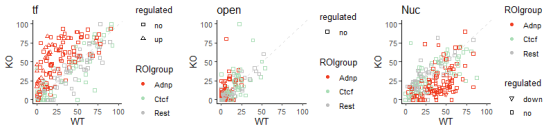
这样,我们就可以将所有核小体模式片段合并在一起,以便更全面地分析核小体模式的变化。这种方法可以简化分析过程,同时提供更直观的核小体模式变化视图。
通过今天的教程,我们一起探索了如何使用dinoR包来分析ADNP敲除与野生型样本间的染色质足迹差异。希望这些分析方法能为你的研究带来新的洞见。如果你在分析过程中遇到任何难题,记得找小师妹帮忙哦!
无论你是在优化代码,还是在云端进行便捷的分析,云生信神器都能为你提供强大的支持。欢迎试试我们的云生信神器,只需一键上传数据,想要的图就能轻松get~