大家好,我是小果~ 还在为生信分析中FASTA文件的繁琐处理而犹豫不决,甚至头疼不已吗?不用再忍受手动操作的繁琐和耗时了!让小果为大家隆重介绍Seqinr这个强大的R包,它就像一个贴心的助手,专门来拯救你的生信分析之路!Seqinr不仅仅是一个简单的序列分析工具,它以其直观易用的接口和丰富的功能,为我们打开了一个全新的视角来处理和分析序列数据。与传统的Biostrings包相比,Seqinr更加专注于提供用户友好的体验和强大的功能,让你在处理FASTA文件等生物信息数据时事半功倍!所以,如果你正在为生信分析中的序列数据处理而烦恼,不妨试试Seqinr这个R包吧!它一定会让你的分析工作变得更加轻松和高效!同时呢,十年的生信之路,小果已经练就了一身扎实的本领,现在准备用这身本事为小伙伴们服务啦!如果你在生信分析上遇到了难题,那就来找小果吧!小果会用自己的专业知识和技能,为你解决困扰,助你一臂之力。期待你的联系哦~
(一) Seqinr R包简要介绍
Seqinr提供了一套全面的功能,帮助研究人员从DNA、RNA和蛋白质序列中提取有价值的信息。Seqinr不仅支持序列数据的读取和写入,还提供了序列处理、转换和分析的多种函数。
Seqinr能够轻松读取和写入各种格式的序列数据,包括FASTA、GenBank和EMBL等。无论是从本地文件还是从在线数据库,Seqinr都能高效地处理这些数据。并且Seqinr提供了一系列函数,用于序列的编辑、转换和比对等操作。例如,计算序列的碱基组成(包括碱基频率和GC含量)。此外,Seqinr还为R语言提供了访问序列数据库的方式,如GenBank等。这使得我们能够更加方便地检索和获取所需的序列数据。
(二) Seqinr R包代码实操
Seqinr可以通过R语言的包管理器进行安装。安装完成后,加载Seqinr包并调用相应的函数即可开始使用。Seqinr的接口直观易用,即使对于没有丰富编程经验的小白来说,也能够快速上手哦~但是该R包操作会占用内存较大,小果建议使用服务器,欢迎小伙伴们联系小果租赁性价比居高的服务器!
快速上手:
1. 安装与加载:
if (!requireNamespace(“seqinr”, quietly = TRUE)) {
install.packages(“seqinr”)
}
library(seqinr)
2. 读取FASTA文件:
seq_records <- read.fasta(file = “sequence1.fasta”,as.string = TRUE)
protein_sequence <- read.fasta(file = “sequence2.fasta”,as.string = TRUE)
# 查看序列信息
names(seq_records)
summary(seq_records)

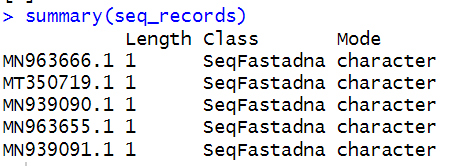
从输出结果我们可以看到,names(seq_records) 函数显示了读取的fasta文件中包含的五个序列的名称,它们分别是 “MN963666.1″、”MT350719.1″、”MN939090.1″、”MN963655.1” 和 “MN939091.1″。这些名称是序列的标识符、访问号,有助于我们在研究中唯一识别每个序列。
summary(seq_records) 函数显示的结果让我们了解到 seq_records是一个包含单个元素的列表,而每个元素又是一个字符模式,代表一个 DNA 序列。每个序列都以它的名字作为标签,存储在列表中。
这些信息对于我们进一步的生物信息学分析非常重要,例如序列比对、变异检测或进化分析。通过这些序列的标识符,我们可以追溯到原始数据源,从而获取更多关于序列的背景信息。
3. 分析序列特性:
# 获取序列长度
sequence_lengths <- c()
# 使用 for 循环获取每个序列的长度
for (i in seq_along(seq_records)) {
sequence_length <- nchar(as.character(seq_records[[i]]))
sequence_lengths <- c(sequence_lengths, sequence_length)
}
# 创建一个数据框来存储序列长度和对应的序列名称
lengths_df <- data.frame(Length = sequence_lengths, Sequence = names(seq_records))
print(lengths_df)
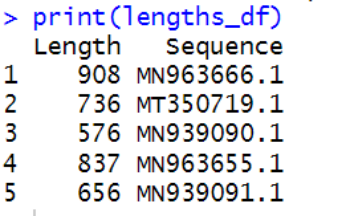
在对提供的FASTA文件 “sequence1.fasta” 进行分析后,我们得到了五个不同的DNA序列,它们分别被标识为MN963666.1、MT350719.1、MN939090.1、MN963655.1和MN939091.1。这些序列的长度从576到908个核苷酸不等,显示出序列间存在一定的长度多样性。序列长度的多样性可能对某些生物学分析有重要影响,例如基因组组装或基因表达分析。
这些序列的具体长度如下:MN963666.1序列长度为908,MT350719.1为736,MN939090.1为576,MN963655.1为837,而MN939091.1为656。序列长度的这种差异可能反映了它们在基因组中的功能多样性或它们所属的基因家族的不同。在进一步的分析中,这些序列可用于比较基因组学研究,以探索它们在进化过程中的保守性和变异性。
# 绘制直方图,为每个序列使用不同的颜色
par(mfrow=c(1,1))
colors <- rainbow(length(seq_records))
# 初始化直方图,不填充颜色
hist(lengths_df$Length, col=”white”, main=”Sequence Length Distribution”, xlab=”Sequence Length”, freq=TRUE, border=”white”)
for (i in seq_along(seq_records)) {
hist(lengths_df$Length, col=colors, add=TRUE, freq=TRUE)
}
在分析序列长度分布时,我们使用了直方图来可视化不同序列的长度。通过rainbow函数为每个序列分配不同的颜色,增强了图表的视觉区分度。更清晰地展示序列长度的多样性和分布特征。
# 计算 GC 含量
calculate_gc_content <- function(sequence) {
g_count <- sum(unlist(strsplit(tolower(sequence), “”)) == “g”)
c_count <- sum(unlist(strsplit(tolower(sequence), “”)) == “c”)
seq_length <- nchar(sequence)
if (seq_length > 0) {
return((g_count + c_count) / seq_length)
}
else {
return(NA)
}
}
gc_content_list <- numeric(length(seq_records))
for (i in seq_along(seq_records)) {
gc_content_list[i] <- calculate_gc_content(seq_records[i])
}
# 打印每个序列的 GC 含量百分比
for (i in seq_along(gc_content_list)) {
sequence_name <- names(gc_content_list)[i]
gc_percentage <- gc_content_list[i] * 100
cat(paste0(“GC content of sequence “, sequence_name, “: “, gc_percentage, “%\n”))
}

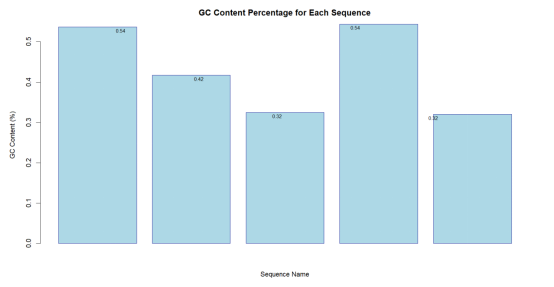
从输出结果可以知道,在对五个DNA序列进行分析后,我们计算了它们的GC含量(即Guanine和Cytosine的比例),结果显示这些序列的GC含量存在显著差异。具体来看,序列的GC含量范围从32.01%到54.36%不等,表明它们在碱基组成上具有多样性。
较高的GC含量通常与基因表达的稳定性和蛋白质编码能力有关,而较低的GC含量可能与基因组中的非编码区域或基因间的间隔区域有关。这种GC含量的多样性可能反映了不同序列在生物学功能和进化压力上的不同适应性。
分析结果表明,其中两个序列具有超过50%的GC含量,这可能指示它们在结构稳定性和功能上的重要性。而其他三个序列的GC含量较低,可能在基因表达调控或基因组结构中有其他作用。这些数据对于进一步的基因组学研究和理解序列的生物学特性具有重要意义。
#可视化GC含量差异情况
par(mar = c(5 ,5, 3, 3))
barplot(height = gc_content_list,
names.arg = names(gc_content_list),
main = “GC Content Percentage for Each Sequence”,
xlab = “Sequence Name”,
ylab = “GC Content (%)”,
col = “lightblue”,
border = “darkblue”)
# 在条形图上添加数值标签
for (i in seq_along(gc_content_list)) {
label <- sprintf(“%.2f”, gc_content_list[i])
text(x = i, y = gc_content_list[i] + 0.01 * max(gc_content_list),
labels = label, pos = 1, offset = 0.5, cex = 0.7)}
# 获取序列名称,使用序列在列表中的索引作为名称
sequence_names <- names(seq_records)
# 初始化一个空列表来存储每个序列的碱基计数结果
base_counts_list <- list()
# 遍历每个序列,计算碱基计数
for (i in seq_along(seq_records)) {
sequence <- seq_records[[i]]
split_sequence <- strsplit(sequence, “”)[[1]]
base_counts <- table(split_sequence)
base_counts_list[[i]] <- base_counts
}
# 将列表转换为数据框
base_counts_df <- do.call(rbind, base_counts_list)
rownames(base_counts_df) <- sequence_names
# 打印结果
print(base_counts_df)
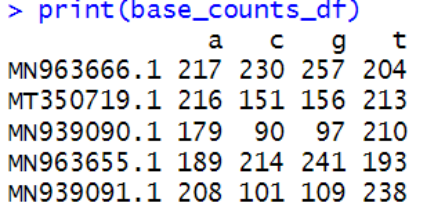
根据结果显示,五个DNA序列的碱基组成各有特点。序列MN963666.1具有最高的G碱基数量(257),而序列MN939090.1的C碱基数量相对较少(90)。整体来看,各序列的A和T碱基数量相对均衡,但G和C碱基的数量在不同序列间变化较大。这些差异可能与序列的功能、结构稳定性以及它们在基因组中的位置有关。GC含量较高的序列可能在热稳定性和基因表达上具有特定的生物学意义。此外,碱基组成也可能影响DNA的三维结构和与其他分子的相互作用。
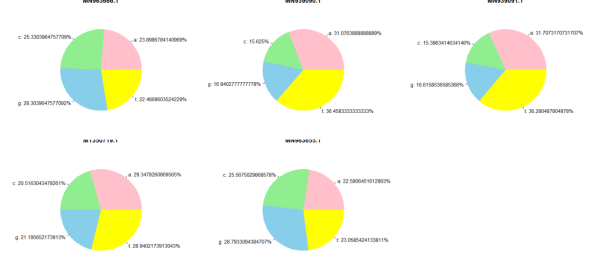
#可视化每条序列的碱基组成情况
unique_bases <- names(base_counts)
base_colors <- colorRampPalette(c(“pink”, “lightgreen”, “skyblue”, “yellow”))(length(unique_bases))
draw_pie <- function(data, name) {
percentages <- data/sum(data) * 100
pie(data, labels = paste0(unique_bases, “: “, percentages, “%”),
main = name, col = base_colors,
border = NA)
}
# 使用layout函数设置画布布局,这里设置为2行3列
layout(matrix(1:6, nrow = 2, ncol = 3))
# 绘制每个序列的饼图
for (i in 1:nrow(base_counts_df)) {
par(mar = c(0, 0, 0, 0))
draw_pie(base_counts_df[i, ], rownames(base_counts_df)[i])
}
# 计算序列长度
sequence_length <- nchar(protein_sequence)
print(sequence_length)

#进行序列检索
sequence_name <- “MN963666.1”
specific_sequence <- as.character(seq_records[[sequence_name]])
print(specific_sequence)

#特殊序列查询
pattern <- “agtc”
matches <- grep(pattern, as.character(specific_sequence))
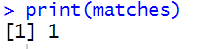
在对特定序列 specific_sequence 进行模式匹配分析时,我们使用了 grep 函数来搜索子串 “agtc”。结果显示,该模式在序列中出现了一次,其位置索引为1。这表明 “agtc” 子串在序列中至少有一个确切的匹配。
分析这一结果,我们可以推断 “agtc” 可能是该序列中的一个保守区域或功能单元。这种模式的出现可能与序列的生物学功能或结构特征相关。虽然仅发现一次匹配,但这足以引起注意,特别是在寻找基因调控元件或蛋白质结合位点时。
进一步的分析可能需要考虑序列的上下文信息,以及 “agtc” 模式在序列中的确切位置和分布情况。这种分析对于理解序列的生物学意义和设计后续的实验验证是有价值的。
#翻转序列并且进行文件的保存输出
reversed_seq <- rev(specific_sequence)
write.fasta(reversed_seq, “path_to_output_file.fasta”)
(三) 文章小结
本期小果和小伙伴们一起进行了Seqinr包的学习, 通过Seqinr包,我们能够更高效地处理和分析序列数据,为未来的科研工作打下了坚实的基础。希望这次学习不仅能让小伙伴们领略到生物数据分析的强大魅力,还能深刻感受到科学探索的无限可能。让我们携手前行,继续在生物信息学的道路上探索未知,追寻真理!最后如果各位小伙伴们觉得自己运行代码太麻烦,欢迎用我们的云生信小工具,只要输入合适的数据就可以直接出想要的图呢,附云生信链接(http://www.biocloudservice.com/home.html)。