大家好~我是小果!不知道小伙伴们是否和小果一样对生物的起源和发展好奇呢?它们是如何从远古时期演化至今,形成了今天我们所见到的形态各异、功能独特的生物种类?今天,和小果一起探索这亿万年的生物演变历程,揭开生物进化的神秘面纱。小果本期将使用ggtree和ape这两个强大的工具,它们不仅能够绘制出令人惊叹的生物进化树,还能帮助我们深入理解生物间的亲缘关系和演化路径。不论你是对生物信息学充满热情的新手,还是已经在这个领域有着丰富经验的专家,都不要错过这次深入了解生命奥秘的机会。让我们用ggtree和ape这两个工具,开启探索生物进化的大门,体验亿万年间生物演变的壮丽史诗,共同感受生命之树的无限魅力!快和小果一起踏上这场激动人心的探索之旅,发现生命的无限可能,领略自然界的奇妙和伟大!同时呢,十年的生信之路,小果已经练就了一身扎实的本领,现在准备用这身本事为小伙伴们服务啦!如果你在生信分析上遇到了难题,那就来找小果吧!小果会用自己的专业知识和技能,为你解决困扰,助你一臂之力。期待你的联系哦~
一、 ggtree与ape R包简要介绍
- ggtree R包介绍
ggtree是ggplot2的拓展包,专门用于绘制生物进化树并进行丰富的注释分析。这一工具由香港大学余光创博士编写,旨在通过图层化的语法来绘制复杂的进化树图形。与ggplot2类似,ggtree允许用户通过组合不同的图层来创建高度定制化的图形。
- 核心功能及特点:
- 广泛的文件格式支持:ggtree能够读取并转换多种软件(如BEAST、r8s、RAxML等)所生成的数据格式,包括Newick、Nexus等。
- 丰富的注释功能:除了基本的进化树绘制外,ggtree还支持在树上添加丰富的注释信息,如节点标签、分支长度、物种名称等。
- 高度的灵活性:用户可以通过调整参数来定制图形的外观和布局,如树状图的形状(矩形、扇形等)、开放角度等。
ggtree自发布以来受到了国际同行的广泛赞誉。相关论文在英国生态学会的Methods in Ecology and Evolution发表后,在不到一年的时间里引用次数已经超过100次,显示出 ggtree R包受欢迎程度和影响力。
- ape R包介绍
APE(Analysis of Phylogenetics and Evolution)是一个用于分子进化和系统发育分析的免费R软件包。它提供了多种用于处理和分析系统发育树的函数和方法,涵盖了从计算进化距离、重建基因树到比较数据分析、进化速率估计等多个方面。
- 核心功能及特点:
- 强大的数据处理能力:APE能够读取和写入多种数据格式的系统发育树,并支持多种类型的进化分析。
- 丰富的统计方法:APE提供了多种用于比较和群体遗传分析的高级统计方法,如距离矩阵计算、祖先状态重建等。
- 易于扩展:APE基于R语言的开源框架,用户可以根据自己的需要编写额外的函数和脚本来扩展其功能。
l ape R包作为分子进化和系统发育分析领域的重要工具,在科研界有着广泛的应用和深远的影响。它为我们提供了一个强大而灵活的平台来探索生物之间的进化关系。
二、 代码实操
小果将带小伙伴们进行代码实战:绘制你的进化树,只需几行代码,你就能轻松读取数据、构建并可视化你的进化树。ggtree与ape包的结合,将会让这一过程变得轻松愉快~本次介绍的R包操作会占用内存较大,小果建议使用服务器,欢迎小伙伴们联系小果租赁性价比居高的服务器!
- 安装和加载所需的包:首先检查ggtree和ape包是否已安装,如果未安装则进行安装,并加载这两个包以便使用
if (!requireNamespace(“ggtree”, quietly = TRUE)) {
install.packages(“ggtree”)
}
library(ggtree)
if (!requireNamespace(“ape”, quietly = TRUE)) {
install.packages(“ape”)
}
library(ape)
- 树文件的读入
setwd(“”D:/A)
tree1 <- read.tree(“D:/A/tree1.nwk”)
tree2 <- read.tree(“D:/A/tree2.nwk”)
tree3 <- read.tree(“D:/A/tree3.nwk”)
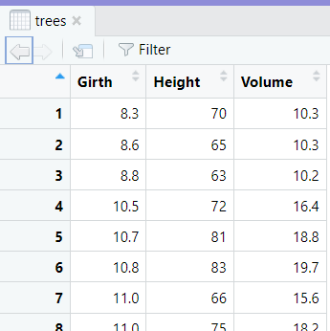
- 树的可视化:使用ggtree函数结合geom_tiplab和geom_nodepoint函数来绘制树,并为叶节点标签和节点点设置颜色和大小。为每棵树设置不同的主题和标题。
p1 <- ggtree(tree1) +
geom_tiplab(aes(color = “orange”), size = 3) +
geom_nodepoint(color = “skyblue”, size = 3) +
theme_tree2() +
ggtitle(“Phylogenetic Tree1 with Branch Lengths”)
print(p1)
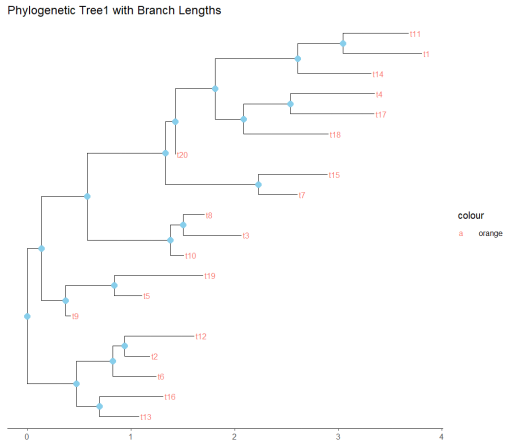
绘制的进化树通过一系列节点展示了t1~t20之间的进化关系。每个节点代表一个分类单元,表示不同种的物种。节点通过颜色编码进行区分,代表不同的进化支或群体。小果利用颜色编码,有助于识别不同的进化群体。
p2 <- ggtree(tree2) +
geom_tiplab(aes(color = “yellow”), size = 5) +
geom_nodepoint(color = “green”, size = 3) +
theme_tree2() +
ggtitle(“Phylogenetic Tree2 with Branch Lengths”)
print(p2)
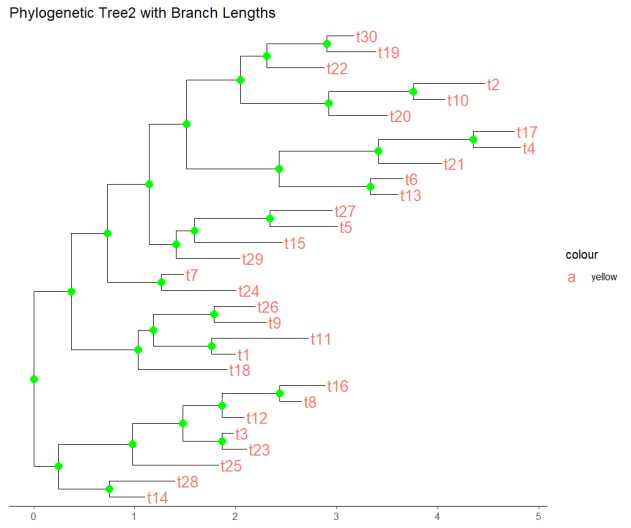
绘制的进化树通过一系列有序的节点,如t2至t30,描绘了从共同祖先分化出的物种间的进化路径。每个节点代表一个特定的分类单元,对应一个物种或物种群。颜色编码,为我们识别不同的进化支提供了直观的视觉区分理解物种间的亲缘关系。通过这些颜色,我们可追踪特定特征或适应性在进化过程中的分布。
p3 <- ggtree(tree3) +
geom_tiplab(aes(color = “red”), size = 4) +
geom_nodepoint(color = “blue”, size = 3) +
theme_tree2() +
ggtitle(“Phylogenetic Tree3 with Branch Lengths”)
print(p3)
绘制出的进化树通过一系列节点(标识为t10至t25以及特异的t2和t1)精细刻画了生物分类单元的系统发育架构。每个节点象征着一个独特的生物分类实体,可能代表单一物种或其亚种群。小果应用颜色编码,这样可以为视觉辨识提供辅助,科学划分不同的系统发育支系。这种编码策略可以揭示出物种间的亲缘关系和潜在的进化路径。
- 添加分支长度标签:使用geom_text函数在p1对象上添加分支长度标签。,因为branch.length直接在aes()函数中创建。
P1 <- p1 + geom_text(aes(label = branch.length), vjust = -1)
print(P1)
和小果一起看看结果吧!
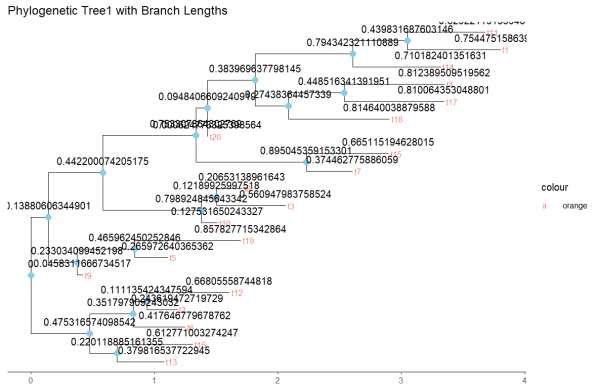
这棵系统发育树以精确的分支长度为特征,展示了从共同祖先演化而来的生物分类单元间的相应的复杂关系。分支长度(如0.439831687603146至0.810064353048801)量化了物种间遗传差异的程度,反映了它们在进化历程中的相对时间距离。节点t17和t18标识了特定的进化分支,这里小果依旧使用了颜色编码为区分不同的进化支或生态位提供了直观的分类工具。
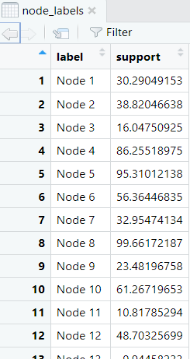
- 节点大小根据支持度调整:小果在代码中创建了一个名为node_sizes的向量,根据支持度调整节点大小,添加节点大小根据支持度
node_sizes <- 2 + (node_labels$support / max(node_labels$support)) * 10
p1 <- p1 + geom_nodepoint(aes(size = support), color = “black”, size = 3)
- 图例添加:添加一个图例,使用是geom_text。
legend_title <- “Branch Length Legend”
p1 <- p1 + geom_text(aes(label = legend_title), x = 0.8, y = 0.7, size = 12)
- 调整布局为圆形: 调整布局为圆形
p1 <- ggtree(tree1, layout = “circular”)
print(p1)
- 计算节点深度和生成节点标签:计算了tree2的节点深度,并生成了一个包含标签和随机支持度的数据框node_labels。
node_depths <- node.depth(tree2)
node_labels <- data.frame(
label = paste(“Node”, 1:length(node_depths), sep = ” “),
support = runif(length(node_depths)) * 100 )
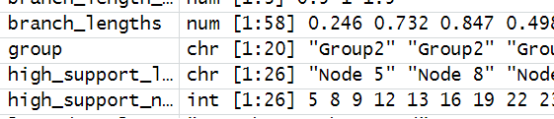
- 分支长度标签添加到第一个树:这部分代码为tree2添加分支长度标签,branch_length在aes()中定义。
branch_lengths <- tree2$edge.length
P1 <- ggtree(tree2, aes(branch.length = branch_lengths)) +
geom_text(aes(label = branch_length), vjust = -1) +
theme_minimal()
- 查找支持度大于50的节点并打印标签:找出node_labels中支持度大于50的节点,并打印这些节点的标签。
high_support_nodes <- which(node_labels$support > 50)
high_support_labels <- node_labels$label[high_support_nodes]
- 突出显示支持度大于50的节点:引用node_labels中的数据创建一个新的树对象P4来突出显示支持度大于50的节点。
print(high_support_labels)

P4 <- p1 + geom_nodepoint(aes(size = support), color = “red”, size = 3)
print(P4)
- 保存图像
ggsave(“Phylogenetic_Tree2_with_Node_Labels_and_Support.png”, p2, width = 10, height = 8, dpi = 300)
ggssave(“Phylogenetic_Tree1.png”, p1, width = 10, height = 8)
ggsave(“Phylogenetic_Tree2.png”, p2, width = 10, height = 8)
ggsave(“Phylogenetic_Tree3.png”, p3, width = 10, height = 8)
三、 文章小结
本期小果和小伙伴们一起利用ggtree和ape包,精心打造了几棵系统发育树。整体而言,这几棵树提供一个关于物种多样性和进化历史的宏观视角。本期利用ggtree包,我们为每棵树添加了色彩和细节,通过geom_tiplab和geom_nodepoint函数为叶标签和节点点赋予了生动的视觉表现。我们还为分支长度标注了标签,并定制了图例,使信息传达更为清晰。为了提升节点的可视化效果,我们创建了一个包含支持度信息的数据框node_labels,并根据支持度动态调整了节点大小,让树的结构更加直观。最后,我们保存了这些精心制作的图像,记录下这次探索的成果。ggtree与ape包是探索生物进化的得力助手,这两个R包让我们能够从微观到宏观,深入解析生命的奥秘。如果你对生物信息学充满兴趣,或渴望更深刻地理解生命演变,不妨亲自尝试这些工具,开启你的发现之旅!最后如果各位小伙伴们觉得自己运行代码太麻烦,欢迎用我们的云生信小工具,只要输入合适的数据就可以直接出想要的图哦,附云生信链接(http://www.biocloudservice.com/home.html)。