前言
小果见面会又要开始了,虽然小果实验很忙,但是看到好用的单细胞聚类R包还是忍不住第一时间向大家分享!想必大多数小伙伴们的细胞类型标记物由聚类后计算的差异基因来鉴定的吧!是不是有时候单细胞类型鉴定和解释的准确性欠佳?不妨跟着小果来看看CASCC这个共表达辅助单细胞聚类方法吧。CASCC的一个关键特点是使用了一种新的自适应吸引子算法,该算法有助于正确的特征选择、聚类和细胞类型鉴定,从而与现有方法相比提高了生物学准确性。CASCC优于其他方法,这已经多个评估指标得到证明啦!所以小伙伴们放心使用哦!没有时间学习的小伙伴们也不要着急哦!有需要生信分析的小伙伴们也可以找小果哦!练了十年生信分析的小果对于生信分析知识已经如鱼得水从分析到可视化直到你满意为止!
单细胞数据处理起来占用内存实在太大了,放过自己的电脑吧!小果在这里给大家送上福利了,有需要服务器的小伙伴们,欢迎大家联系小果,保证服务器的性价比最高哦!
实战教程
1.安装CASCC
1.1使用anaconda安装R和CASCC
conda create -n CASCC_env
source activate CASCC_env
conda install conda-forge::r-fs # devtools是必备的
conda install conda-forge::r-devtools
R
.libPaths(“./anaconda3/envs/CASCC_env/lib/R/library”) # 设置conda R库路径
install.packages(“rlang”,version=”1.1.0″) # 依赖包:更新rlang包
remotes::install_version(“Seurat”, “4.3.0”) # 依赖包
devtools::install_github(“weiyi-bitw/cafr”) # 依赖包
devtools::install_github(“LingyiC/CASCC”)
CASCC::checkDependencies()
1.2选项2:在R中安装
require(devtools)
remotes::install_version(“Seurat”, “4.3.0”) # dependency
install_github(“weiyi-bitw/cafr”) # dependency
install_github(“LingyiC/CASCC”)
CASCC::checkDependencies()
2. CASCC使用教程
在本教程中,我们将分析来自(Peng et al. 2019)研究的胰腺导管腺癌(PDAC)患者的示例数据集。这个例子包括8种不同的细胞类型。
2.1快速入
2.1.1用一行代码运行CASCC
这里我们使用main函数run.CASCC来运行CASCC。逐步运行CASCC请参见“逐步运行CASCC”一节。
rm(list = ls()); gc()
library(CASCC)
library(Seurat)
set.seed(1)
## 加载示例数据
data(“Data_PDAC_peng_2k”)
dim(Data_PDAC_peng_2k) # 400 cells
## 运行CASCC
CASCC::checkDependencies()
##inputDataType ‘ data ‘被规范化和对数转换。
res <- CASCC::run.CASCC(Data_PDAC_peng_2k)
labels <- res$mainType.output$clusteringResults
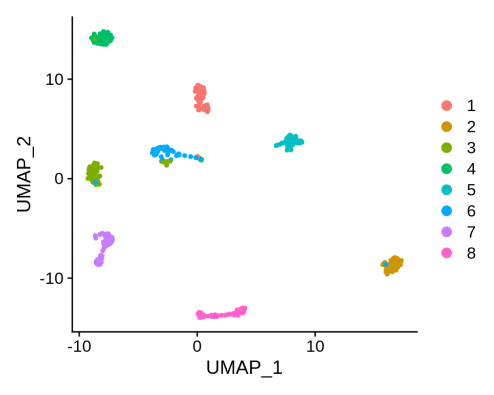
2.1.2UMAP可视化
## 绘图
adata <- res$res.predictK$adata # UMAP
labels <- res$mainType.output$clusteringResults
# 聚类输出:具有吸引子中心的k均值
Idents(adata) <- labels
DimPlot(adata)
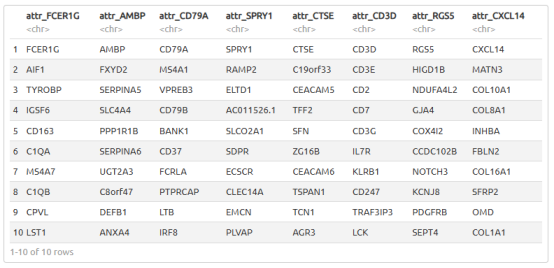
2.1.3吸引子共表达式输出
分析确定了八个不同集群的存在,每个集群对应一个特定的吸引子,如下表所示。我们展示了每个吸引子的前10个基因,它们与已知的细胞类型标记密切相关。从左到右依次为巨噬细胞、1型导管细胞、B细胞、内皮细胞、2型导管细胞、T细胞、星状细胞和成纤维细胞。
## 结果列表
attr.raw <- res$fs.res$attr.raw
# 原始的吸引器扫描结果,如果重新运行CASCC可以使用。
# res$fs.res$attrs # 删除相同的吸引子
finalAttrs <- res$fs.res$finalAttrs # 过滤吸引子
## 吸引子用作聚类中心
attrs <- res$mainType.output$attrs.center
df <- as.data.frame(do.call(cbind, lapply(attrs, function(x) {data.frame(“gene” = names(x))})))
titles <- c()
for (attr.ID in paste0(“attr_”, unlist(lapply(attrs, function(x){names(head(x)[1])})))) {
titles <- c(titles, c(attr.ID))
}
colnames(df) <- titles
df[1:10, ] # 每个吸引子的前10个基因(行)(列)
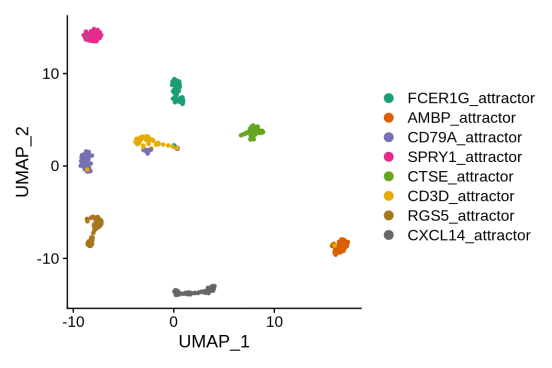
2.1.4 UMAP,由吸引子标记
将确定的8个吸引子用作每个亚簇的中心。
Idents(adata) <- res$mainType.output$clusteringResults
nClust <- length(unique(Idents(adata)))
mycolor <- grDevices::colorRampPalette(RColorBrewer::brewer.pal(n = 8, name = “Dark2”))(nClust)
attractorDimplot(adata, attr.list = res$mainType.output$attrs.center, col = mycolor)
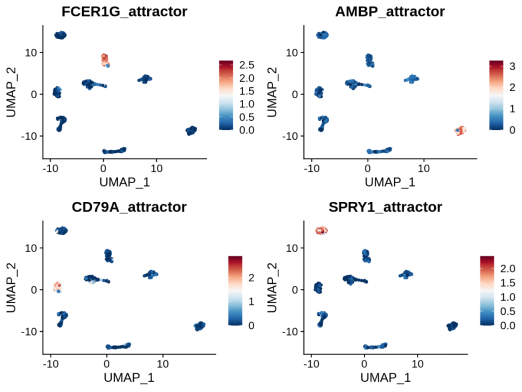
2.1.5 Attractor plot
每个吸引子中前5个基因的平均表达量。
attractorPlot(adata, attrs[1:4], n.col = 2)
2.1.6寻找差异表达基因
我们发现亚群之间存在差异表达基因(DEGs)。
DEGs <- Seurat::FindAllMarkers(adata, only.pos = T)
head(DEGs
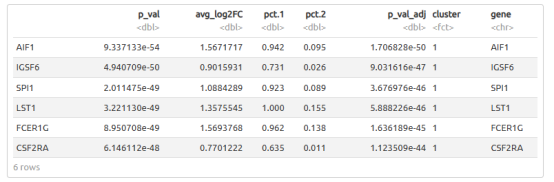
2.2快查找共表达特征基因
我们提供了一种自适应版本的吸引子算法,基于所提供的种子基因来寻找收敛的吸引子。如果用户只对共同表达式吸引器感兴趣,则可以使用以下代码查找共同表达式,而无需运行整个CASCC管道。
library(“CASCC”)
library(“cafr”)
data(“Data_PDAC_peng_2k”)
# 找到AIF1种子吸引子。AIF1基因是巨噬细胞的标志物。
attr.res <- findAttractor.adaptive(Data_PDAC_peng_2k, “AIF1”)
# 共表达的吸引子
head(names(attr.res$attractor.final))
#> [1] “FCER1G” “AIF1” “TYROBP” “IGSF6” “CD163” “C1QA”
2.3逐步运行CASCC
在本节中,我们将逐步展示如何运行CASCC。
rm(list = ls()); gc()
library(“CASCC”)
data(“Data_PDAC_peng_2k”)
# 1-3步
fs.res <- CASCC.featureSelection(Data_PDAC_peng_2k, inputDataType = “well-normalized”, attr.raw = NULL, exponent.max = 10, exponent.min = 2,
generalSeedList = NULL, mc.cores = 1, topDEGs = 10, topAttr = 50,
removeMT.RP.ERCC = TRUE, removeNonProtein = FALSE,
topN.DEG.as.seed = 1,
overlapN = 10)
adata <- fs.res$adata;
SeuratVersionCheck = as.numeric(strsplit(as.character(packageVersion(“Seurat”)), “\\.”)[[1]][1])
if (SeuratVersionCheck == 4) {
data <- adata[[“RNA”]]@data
}else if (SeuratVersionCheck == 5) {
data <- adata[[“RNA”]]$data
}
data <- as.matrix(data)
#第四步
res.predictK <- predictClusterK(data, fs.res, method = “ward.D2”, index = “silhouette”, min.nc_fix = FALSE)
# 结果列表
res <- list()
res$fs.res <- fs.res
res$res.predictK <- res.predictK
# 第五步
res$mainType.output <- kmeansCenterModify(res)
3.其他脚本
3.1生成示例数据集
set.seed(1)
rm(list = ls()); gc()
## 原始数据来自于 PRJCA001063
load(“./adata.T15.RData”)
load(“./cell.type.RData”)
cell.type <- cell.type[which(cell.type$cell.name %in% colnames(adata)), ]
table(cell.type$cluster)
library(dplyr)
sampled_cells <- cell.type %>%
group_by(cluster) %>%
## if a cell type does not have more than 50 cells, we removed this cell type
sample_n(size = if_else(n() >= 50, 50, 0)) %>%
ungroup()
print(sampled_cells)
genes <- Seurat::VariableFeatures(adata)
cells <- sampled_cells$cell.name
SeuratVersionCheck = as.numeric(strsplit(as.character(packageVersion(“Seurat”)), “\\.”)[[1]][1])
if (SeuratVersionCheck == 4) {
data <- as.matrix(adata[[“RNA”]]@data[genes, cells])
}else if (SeuratVersionCheck == 5) {
data <- as.matrix(adata[[“RNA”]]$data[genes, cells])
}
save(data, file = “./CASCC/data/Data_PDAC_peng_2k.RData”)
start_time <- Sys.time()
res <- CASCC::run.CASCC(data)
end_time <- Sys.time()
end_time – start_time
save(res, file = “./CASCC/data/Res_PDAC_peng_2k.RData”)
3.2在Seurat工作流中运行CASCC
这里我们将展示如何在Seurat工作流中运行CASCC。
SeuratVersionCheck = as.numeric(strsplit(as.character(packageVersion(“Seurat”)), “\\.”)[[1]][1])
if (SeuratVersionCheck == 4) {
data <- SeuratObject[[“RNA”]]@data
}else if (SeuratVersionCheck == 5) {
data <- SeuratObject[[“RNA”]]$data
}
res <- CASCC::run.CASCC(data)
Idents(SeuratObject) <- res$mainType.output$clusteringResults
SeuratObject
小结
今天小果带着大家学习了一种用于scRNA-seq分析的共表达辅助单细胞聚类(CASCC)方法。CASCC的一个关键特点是使用了一种新的自适应吸引子算法,该算法有助于正确的特征选择、聚类和细胞类型鉴定,从而与现有方法相比提高了生物学准确性。最后小果给大家介绍一个云工具!同学们如果觉得自己的代码水平一般,对于很多的参数不知道怎么改,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表,字体大小、标题等也可一键更改。感兴趣的小伙伴去云生信(http://www.biocloudservice.com/home.html)体验一下吧!