前言
你是不是还在为了建立遗传变异与疾病结果之间错综复杂的关系而发愁?别担心,小果给大家找了一个超级好用的python库,分析、可视化一个包全部搞定!今天,小果给大家介绍的工具就是Genopyc,这是一个新颖的Python库,旨在全面研究与复杂疾病相关的变异如何影响下游途径。Genopyc为异构数据挖掘和可视化提供了一套广泛的功能,使研究人员能够深入研究和整合来自大规模基因组数据集的生物信息。为了更好地给大家介绍如何使用该工具,小果给大家找了一个示例数据向大家手把手教学。下面小果带着大家使用 Genopyc 研究与椎间盘退行性变相关的变异的功能后果,更深入地了解该疾病中涉及的潜在失调通路,并利用软件包中的功能进行探索和可视化。练了十年生信分析的小果对于生信分析知识已经如鱼得水,相信有小果的加持一定能让大家学会!没有时间学习且有生信分析的小伙伴们快来滴滴小果噢!从分析到可视化直到你满意为止!
小果在这里给大家送福利了,有需要服务器的小伙伴们,欢迎大家联系小果,保证服务器的性价比最高哦!
实战教程
在本教程中,小果将在研究椎间盘退变(IDD)中教给大家如何使用该python库,这是椎间盘突出和背部疼痛的主要原因之一的复杂特征。我们将首先通过全基因组关联研究(GWAS)收集与该性状相关的snp,并阐明哪些途径受到变异的干扰。
安装相关python库
#!pip install genopyc
#!pip install graphlot
导入所有必要的库
import genopyc as gp
import matplotlib.pyplot as plt
import pandas as pd
import ast
import matplotlib_venn as pltv
import networkx as nx
import graphlot as gv
from upsetplot import from_contents
from upsetplot import UpSet
从GWAS目录中下载与椎间盘退变相关的变异(EFO_0000305)
##这一部分数据下载有困难的小伙伴记得私信小果获取数据!!!
associations = pd.read_csv(‘./data/EFO_0004994_associations_export.tsv’,sep=’\t’)
#分离2中的riskAllele列并删除该列
associations[[‘rsid’, ‘RiskAllele’]] = associations[‘riskAllele’].str.split(‘-‘, expand=True)
associations.drop(‘riskAllele’,inplace=True,axis=1)
#将rsid设置为索引
associations.set_index(‘rsid’,inplace=True)
associations
##或者,可以使用get_associations()函数直接从GWAS编目中检索关联。函数接受trait的efo代码作为参数。
%%time
gp.get_associations(‘EFO_0004994’,verbose=True)
#查询EFO_0004994的关联关系…
#构建数据框架…
#CPU时间:user 41.4 ms, sys: 205µs, total: 41.6 ms
#开机时间:2.35秒
得到变异的基因组坐标
我们将在2个变量中存储snp列表和变体的位置,以便与它们一起工作。位置可以从关联数据框中获取,也可以使用函数get_variants_position()获取,该函数接受一组rsid作为参数。在这种情况下,我们将使用关联数据框中已经存在的坐标。
variants=list(set(associations.index.tolist()))
variants[:10]
variant_positions=[(i,r[‘locations’].split(‘:’)[0],r[‘locations’].split(‘:’)[1]) for i,r in associations.iterrows()]
variant_positions[:10]
gp.get_variants_position(variants)[:10]
获取变量列表的规则元素
第一种方法可能是研究存在于感兴趣的变体周围的特定区域的元素。这可以通过get_ov_elements()函数轻松完成。它接受基因组坐标染色体,特定基因组区域的起始和结束并返回重叠的元素。feature关键字是一个包含我们想要检索的特定特性的列表。请参阅来自ensembl的文档,以获得可以检索的可能特性的完整列表。
函数get_ov_region有一个模式SNP,该函数在其中接受单个遗传变异作为输入,还有一个模式区域,其中可以传递基因组区域(染色体、起始、终止)。
下面的例子展示了如何检查rsid rs2901157周围500 bp的区域,这将返回该基因组区域中的基因和基序特征
overlapping_elements = gp.get_ov_region(‘rs1981483’,features=[‘regulatory’,’gene’,’motif’],mode=’SNP’,window=200)
overlapping_elements[2]
结果是我们查找的每个特性类型的数据框列表。例如,我们可以检索该区域的所有转录因子结合位点。一种有趣的方法可能是研究给定变异的表达数量性状位点(eqtls)。这些研究将遗传变异与基因表达的改变联系起来,更有可能的是,SNP对特定候选基因的表达有影响。
通过Eqtls连接变异与基因
Genopyc允许您检索给定代码变体的所有等价项。数据从EBI中获取。在这里,我们将得到变量的等式。该函数可能需要一段时间才能运行
eqtls_dict={rsid:gp.get_eqtl_df(rsid) for rsid in variants}
eqtls_dict={rsid:gp.get_eqtl_df(rsid) for rsid in variants}
为了更好地可视化数据,我们可以构建一个异构网络
list_of_eqtls_dataframes = list(eqtls_dict.values())
eqtl_tissues = set(list(sum([d.tissue_name.tolist() for d in list_of_eqtls_dataframes],[])))
eqtl_tissues
通过使用make_eqtl_network()函数,我们将构建一个变异-基因-组织的网络
该网络将帮助我们更好地理解变异的作用事实上,如果一个变异抑制特定基因的转录,红色边缘将显示给我们,而如果变异增强转录,绿色边缘将显示给我们。我们将过滤骨骼肌组织的结果以便将注意力集中在这个特定的组织上。
eqtl_network_smt = gp.make_eqtl_network(list_of_eqtls_dataframes,tissue_name=True,gene_symbol=True,tissue=’skeletal muscle tissue’)
结果将是一个网络,其中节点是变体基因或组织,如果变体抑制或增强基因的表达,则变体和基因之间存在边缘。如果基因在特定组织中表达,则基因和组织之间存在边缘。
通过使用graphlot库,我们将对骨骼肌组织的过滤结果进行可视化。
graphlot库是一个灵活的网络可视化工具。它允许我们在2d和3d模式下可视化网络,并根据在我们的eqtl_network中编码的数据来定义节点和边缘。这里的绿色边缘表示转录增强,而红色表示抑制。宽度与变异对基因转录的影响成正比。
gv.visualize_network( eqtl_network_smt,mode=’2d’,
layout=’auto’,
edge_annotation_attributes=[‘beta’],
edge_color_attribute=’color’,
node_color_attribute=’tipo’,
node_annotation=True,
show_node_label_3d = True,
edge_attribute_width=’beta’,
factor_edge_width=4,
node_size=200,
)
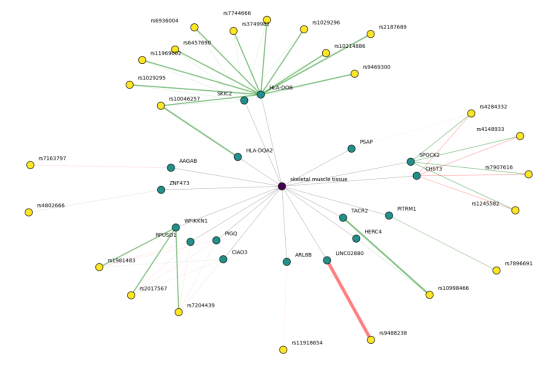
Eqtl是一种将变异与基因连接起来的有趣方式,这里我们将通过eqtls(即转录机制的改变)获取受变异影响的基因,并将它们存储在一个名为eqtl_genes的变量中
eqtl_genes = [node[0] for node in eqtl_network_smt.nodes(data=True) if node[1][‘tipo’] == ‘gene’]
eqtl_genes
运行VEP来调查变异结果,绘制结果并将变异与基因联系起来。
用函数VEP我们可以编程地运行VEP来调查SNP的后果。该函数接受rsid列表或HGVS id。为了达到我们的目的,我们将使用HGVS符号,因为GWAS研究为我们提供了相对于参考和替代等位基因的精确信息。
#从关联数据框中获取风险等位基因
variant_risk_alleles=dict(zip(associations.index.tolist(),[ra for ra in associations.RiskAllele.tolist()]))
# 从关联数据框中获取基因组位置
variant_genomic_locations=dict(zip([tup[0] for tup in variant_positions],[tup[2] for tup in variant_positions]))
variant_chromosomes=dict(zip([tup[0] for tup in variant_positions],[tup[1] for tup in variant_positions]))
#使用get_ancestral_allele函数检索祖先等位基因
variant_ancestral_allele = gp.get_ancestral_allele(variants)
#要合并检索到的所有信息并将其转换为HGVS表示法,请运行:
hgvs_dict = {}
for variant in associations.index.tolist():
try:
hgvs_notation=str(variant_chromosomes[variant])+’:g.’+str(variant_genomic_locations[variant])+str(variant_ancestral_allele[variant])+’>’+str(variant_risk_alleles[variant])
hgvs_dict[variant] = hgvs_notation
except Exception as error:
print(f’Error for rsid {variant}: ‘ + repr(error))
hgvs_dict
这段代码的结果是一个以rsid为键的字典和对应的HGVS表示法变体,我们可以很容易地检索它们之间的映射。
我们可以看到有时风险等位基因和祖先的等位基因是一样的。当替代等位基因对表型有一定的保护作用(负奇数比)时,替代等位基因可能比替代等位基因更频繁或者祖先等位基因是风险等位基因。
我们还可以看到,有时GWAS的风险等位基因被标记为“?”。在这种情况下,我们没有与疾病相关的变异相关的信息。
使用HGVS方法,这些snp将被排除在分析之外。一种可能的方法是在函数gp.VEP()中使用输入type = “rsid”
VEP可以通过传递参数input_type=’hgvs’并作为list函数的参数来使用hgvs表示法运行
vep_consequences_hgvs=gp.variant_effect_predictor(list(hgvs_dict.values()),plot=True,input_type=’hgvs’)
vep_consequences_hgvs[2]
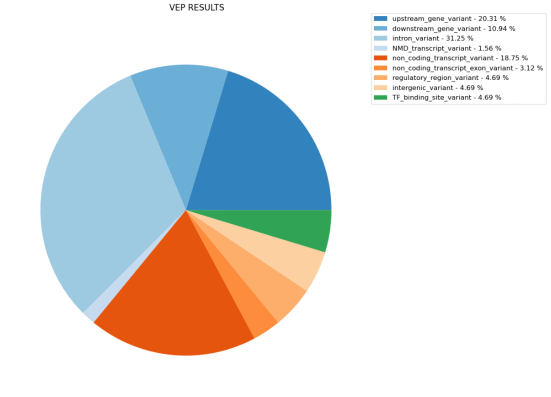
输出是一个包含4个数据帧的列表。VEP将数据分为转录效应、调控效应、非编码效应和调控基序效应。下面,我们展示了列表中的第二个数据框,它存储了所有的调节效果。
将变异与VEP后果的基因联系起来
Vep是我们结合变异和基因的另一种方式,这里我们将创建两个变量:通过转录因子(调节)和编码变异相关的基因。
vep_consequences_hgvs[3]
tflink = pd.read_csv(‘./data/TFLink_Homo_sapiens_interactions_SS_simpleFormat_v1.0.tsv’,sep=’\t’)
tf_affected = vep_consequences_hgvs[3].TF.tolist()
tf_affected = [ast.literal_eval(e) for e in tf_affected]
tf_affected = sum(tf_affected,[])
tf_affected = set(sum([l.split(“::”) for l in tf_affected],[]))
#检索由这些tf调控的基因
genes = []
for tf in tf_affected:
tf_genes = tflink[tflink[“Name.TF”] == tf][“NCBI.GeneID.Target”].tolist()
genes.extend(tf_genes)
## 美化列表(删除重复项和具有多个基因的字符串)
genes_tfbs = list(set(sum([g.split(“;”) for g in [d for d in genes if d != ‘-‘]],[])))
print(f”A total of {len(genes_tfbs)} genes are controlled from the transcription factors”)
受转录因子控制的基因共有870个
由于TFLink不是组织特异性的,因此有许多基因的转录受到受变异影响的TFS的控制。一种可能的方法是将基因集限制在磁盘中实际表达的基因上。我们将使用一项名为Dipper的蛋白质组学研究的数据。
此外,我们还需要在基因标识符(entrez, ensembl, gene symbol…)之间进行转换,为此我们将使用biomapy库
protein_expressed_disc = pd.read_csv(‘./data/dipper_protein_expression.csv’,index_col = 0 )
genes_list = protein_expressed_disc[‘Gene.names’].tolist()
entrez_list =gp.geneId_mapping(genes_list,’symbol’,’entrez’)
entrez_list = [str(g) for g in list(filter(None,entrez_list))]
pltv.venn2([set(entrez_list),set(genes_tfbs)],set_labels=[“EXPRESSED GENES IVD”,”TFBS GENES”])
plt.title(“INTERSECTION BETWEEN EXPRESSED GENES AND TFBS GENES”)
plt.show()
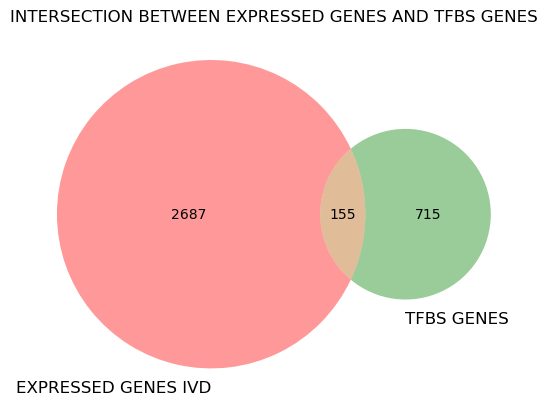
expressed_genes_tfbs = list((set(entrez_list).intersection(set(genes_tfbs))))
expressed_genes_tfbs[:10]
#将它们转换为基因名称
expressed_genes_tfbs_symbol = gp.geneId_mapping([int(a) for a in expressed_genes_tfbs],’entrez’,’symbol’)
expressed_genes_tfbs_symbol[:10]
编码变异
genes_vep = set(vep_consequences_hgvs[1].ENSID.tolist())
print(f”A total of {len(genes_vep)} has been associated through VEP coding consequences”)
##总共有7个与VEP编码结果相关联
genes_vep = list(genes_vep)
genes_vep_symbol = gp.geneId_mapping(genes_vep,’ensembl’,’symbol’)
genes_vep_symbol = list(filter(None,genes_vep_symbol))
genes_vep_symbol
将变异与gwas目录中的基因连接起来
当进行GWAS时,作者通常报告他们发现的每个变异的相关基因。如果变异是基因间的,他们会提到最近的上游和下游基因。该信息在初始数据框的mapappedgenes列中报告。
genes_gwas = list(set(sum([g.split(‘,’) for g in associations.mappedGenes.tolist()],[])))
genes_gwas
利用Open Target L2G管道检索与GWAS位点相关的基因
我们需要做的第一件事是将变体转换为chr_position_reference_alternative等位基因的格式,这可以使用genopyc的variantId_mapping函数来完成。然后我们可以使用OT_L2G函数运行OTG管道。结果是一个包含3列的数据框,其中包含变体id、基因和管道分配的分数。或者,不指定output = ‘all’,将获得列表中得分最高的基因。
variants_to_study_ot = gp.variantId_mapping(variants,source=’rsid’,target = ‘variantid’)
variants_to_study_ot=list(filter(None,variants_to_study_ot))
variants_to_study_ot
该函数接受一个变体列表,输出是L2G管道中得分最高的基因列表。还有一个标志模式,如果等于“all”,则返回管道中具有相关分数的每个基因的数据框
OT_genes = gp.OT_L2G(variants_to_study_ot)
len(OT_genes)
OT_genes_symbol = gp.geneId_mapping(OT_genes,’ensembl’,’symbol’)
OT_genes_symbol到目前为止,我们通过几种方式将变异与基因联系起来,我们可以分析这些方法之间的任何重叠。我们将绘制结果。对于这项任务,我们将使用R的接口,以便使用rpy2接口利用UpSet plot库。
准备可视化的数据
all_genes = list(set(OT_genes_symbol+genes_gwas+expressed_genes_tfbs_symbol+genes_vep_symbol))
gene_categories = [‘OT’,’GWAS’,’TFBS_VEP’,’CODING_VEP’,’EQTLS’]
gene_sets = [ OT_genes_symbol,genes_gwas,expressed_genes_tfbs_symbol,genes_vep_symbol,eqtl_genes]
data = from_contents(dict(zip(gene_categories,gene_sets)))
UpSet(data)
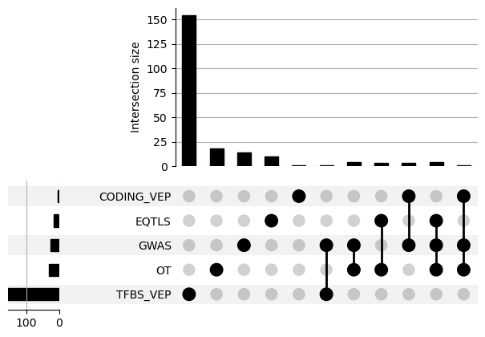
我们可以看到,我们仍然有大量的基因由受变异影响的转录因子控制,但与其他类别几乎没有重叠,这意味着这是一种将变异与基因联系起来的非常非特异性的方式。
另一方面,在GWAS、OT和EQTLS的优先级基因之间存在一簇重叠,这意味着这些方法检测的是相同的基因簇,因此可能与疾病相关。为了研究这些基因的功能和富集途径,我们将对GWAS和OT遗传学中优先排序的一组基因进行功能富集分析。
print(“OTL2G GENES: \n”,OT_genes_symbol,”\n\n”)
print( “GWAS GENES: \n”, genes_gwas)
功能富集分析
def PlotEnrich(gene_list,pval):
gp.plot_enrichment_analysis_network(gene_list,pval,fontsize_ann=30,layout = ‘spring’,colormap=’Blues’,edgecolor=’grey’,
mkcolor=’grey’,figsize=(20,15),k=100,factor=1.1,cbarfontsize=20,mkfsize=2500,
legend=True,legend_fontsize=20,legend_col=1, legend_columnspacing=3,legend_labelspacing=3,
legend_location=(1.5,0.8),legend_title=’Number of Genes’,cbar_loc=(0,0))
PlotEnrich(genes_gwas,0.05)
PlotEnrich(OT_genes_symbol,0.01)
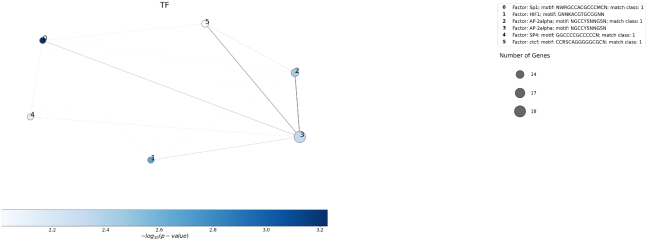
我们可以看到,来自OT L2G管道的基因中富集的功能与文献中已经报道的与IDD发展相关的tf相关。
小结
Genopyc提供了一个多合一的工具来检索和解释基因组变异对复杂疾病发展的影响。Genopyc为异构数据挖掘和可视化提供了一套广泛的功能,使研究人员能够深入研究和整合来自大规模基因组数据集的生物信息。使用 Genopyc 研究与椎间盘退行性变相关的变异的功能后果,可以更深入地了解该疾病中涉及的潜在失调通路,这可以通过利用软件包中的功能进行探索和可视化。Genopyc 可以通过 pip 轻松安装,并且可以集成到基于主要 Python 库构建的 Python 环境中。感兴趣的小伙伴们赶快学起来吧!最后小果给大家介绍一个云工具!同学们如果觉得自己的代码水平一般,对于很多的参数不知道怎么改,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表,字体大小、标题等也可一键更改。感兴趣的小伙伴去云生信(http://www.biocloudservice.com/home.html)体验一下吧!