各位做单细胞的小伙伴们有没有这样的烦恼呀?最近小果总是发现每次都找不到想要往下分析的点,根据现在固有的一些单细胞流程做到细胞分类之后就很难再抓到往下分析的细胞类型了,怎么确定哪个细胞类型是对我们的课题有意义的呢?除了传统的根据细胞比例还确定之外, R包Augur也来帮忙啦!这么一个简单又好用的包,呜呜,简直太称小果的心意了,简单好用,特别好上手,即便是小白也分分钟学得会哦!!!
同时这里小果要提醒大家啦,如果遇到操作占用内存比较大的时候,小果建议大家用服务器哦,不然自己的电脑运行不开,如果没有自己的服务器欢迎联系小果租赁服务器哦~
R包Augur就是用来识别对单细胞数据中的生物扰动最敏感的细胞类型,简单点来说就是比如小果研究的是衰老的课题,假如该数据的样本组成是8个衰老和年轻的某组织的数据,那对于这个课题来说,衰老可易简单理解为这个生物扰动,又比如各种癌症数据等等,我们可以通过这个包计算出受到衰老影响最敏感的一个或几个细胞类型,这样对于我们后续的研究很有帮助哦!当然还有一些疾病治疗的数据,就可以清楚的看到治疗后最关键的细胞类型了!
那这个包当然也不是岌岌无名的,有一些文章就使用了这个包,比如下面这些小果发现的一些使用这个包的一些案例,如下:
案例一:
来源于2023年7月发表在Protein & Cell上的题为A single-nucleus transcriptomic atlas of primate liver aging uncovers the pro-senescence role of SREBP2 in hepatocytes中提到

文章的描述如下:we identified that Hep was the cell type most responsive to aging in the single-nucleus data
是用的单核细胞衰老的数据,计算出了衰老最敏感的一个细胞类型Hep。
案例二:
来源于2023年4月发表在Protein & Cell上的题为Single-nucleus transcriptomics reveals a gatekeeper role for FOXP1 in primate cardiac aging的文章中提到

CM was the cell type most affected by aging
那接下来就跟小果来学习这个包的安装和使用吧~
1.安装
依赖包安装
dplyr (>= 0.8.0),
purrr (>= 0.3.2),
tibble (>= 2.1.3),
magrittr (>= 1.5),
tester (>= 0.1.7),
Matrix (>= 1.2-14),
sparseMatrixStats (>= 0.1.0),
parsnip (>= 0.0.2),
recipes (>= 0.1.4),
rsample (>= 0.0.4),
yardstick (>= 0.0.3),
pbmcapply (>= 1.5.0),
lmtest (>= 0.9-37),
rlang (>= 0.4.0),
glmnet (>= 2.0),
randomForest (>= 4.6-14)
需要提前先安装一下
install.packages(“devtools”)
devtools::install_github(“Bioconductor/MatrixGenerics”)
devtools::install_github(“const-ae/sparseMatrixStats”)
devtools::install_github(“neurorestore/Augur”)
library(Augur)
2.运行
这里使用的实验数据是Augur自带的
data(“sc_sim”)
里面的数据的分类信息如下,一共有两列,第一列是治疗组与control组,第二列就是细胞类型,我们这里就看看哪个细胞类型对于治疗组来说最为敏感。
head(sc_sim@meta.data)
label cell_type
1 control CellTypeA
2 treatment CellTypeA
3 treatment CellTypeA
4 control CellTypeA
5 control CellTypeA
6 treatment CellTypeA
接着运行calculate_auc,并检查AUC项中的细胞类型优先级
augur = calculate_auc(sc_sim)
using default assay: RNA …
|============================================================================================| 100%, Elapsed 01:48
> augur$AUC
# A tibble: 3 × 2
cell_type auc
<chr> <dbl>
1 CellTypeC 0.879
2 CellTypeB 0.747
3 CellTypeA 0.554
>
可以看到我们这里只有三个细胞类型A,B,C,根据优先级,是C>B>A,那可能细胞类型C就是我们需要后续主要关注的一个细胞类型啦!
3.绘图
data <- augur$AUC
names(data)
data$Category <-
paste0(data$cell_type,” – “,round(data$auc,2))
data$Category
[1] “CellTypeC – 0.88” “CellTypeB – 0.75” “CellTypeA – 0.55”
ggplot(data = data, mapping = aes(x = cell_type, y = auc, fill = cell_type)) +
geom_bar(stat = ‘identity’, position = ‘dodge’) +
scale_fill_brewer(palette = ‘Accent’)+#柱形的颜色
ylim(c(0, 1.3))+#y轴的范围,如果不设置,整个图就是一个圆,连起来了,y值越大,图形越短
xlab(“”) + ylab(“”) +
coord_polar(theta = “y”)+# 按照Y轴旋转
geom_text(data = data, hjust = 1, size = 3,
aes(x = Category, y = 0, label = Category)) +#加载X轴上的标签
theme_minimal() +# 去掉灰色背景
theme(legend.position = “none”,
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.line = element_blank(),
axis.text.y = element_blank(),
axis.text.x = element_blank(),
axis.ticks = element_blank())
通过运行上述的代码,我们就可以画出来和文章中一样的图形啦~最后的图形就是下面这样啦!因为示例数据的细胞类型较少,所以做出来的图形不怎么好看,大家可以换自己的数据看看呀,因为一般单细胞的数据细胞类型也是比较多的,基本上都是十几个,这样我们的图形就和文章中的看起来差不多啦!
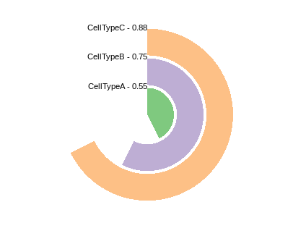
此外还有一些细小的参数小果带大家进行调整,一个是如果大家的细胞数比较多的时候,运行这一步就比较困难了,可以调整n_threads参数,默认下该参数的值是4,我们可以调整成8,这样运行速度就是之前的两倍了。在对接seurat对象时候,meta.data 中具有名为 cell_type 和 label 的列,这意味着我们可以将其直接作为 Augur 的输入提供,输入cell_type_col参数和label_col参数即可。
当然啦,我们用这个包也是为了计算细胞类型的敏感度,至于第三部分绘图的形式,如果大家有其他更合适一点的图形也可以换其他的哦~这里小果只是提供一种展示方式~其实如果细胞类型像小果这种很少的话,可以直接画barplot,就像遇到分析亚类的情况时。
此外Augur寻找细胞类型优先级的原理在github中有给出,Augur is an R package to prioritize cell types involved in the response to an experimental perturbation within high-dimensional single-cell data. The intuition underlying Augur is that cells undergoing a profound response to a given experimental stimulus become more separable, in the space of molecular measurements, than cells that remain unaffected by the stimulus. Augur quantifies this separability by asking how readily the experimental sample labels associated with each cell (e.g., treatment vs. control) can be predicted from molecular measurements alone. This is achieved by training a machine-learning model specific to each cell type, to predict the experimental condition from which each individual cell originated. The accuracy of each cell type-specific classifier is evaluated in cross-validation, providing a quantitative basis for cell type prioritization.小果这里就不再赘述啦!希望这个包能帮助到大家哦~
在如今这个单细胞流行的时代,做单细胞分析似乎已经不可或缺,如果各位朋友还有其他的单细胞的分析的需求,但是平时做实验又没时间学代码的话,可以试试生信分析小工具云平台,这个平台优点就是不需要懂代码,直接将数据输入进去就会出结果啦,真的是代码小白的最爱啦,云平台网址我放在这里啦:(http://www.biocloudservice.com/home.html),像单细胞绘制小提琴图或者tSNE都可以在这里完成哦!!!需要的小伙伴可以去瞧瞧哦~