大家好!今天,我非常激动地向大家介绍生物信息学领域的一项强大工具——Rtsne 包。相信大家都知道,在现代生物信息学研究中,获取和分析生物信息数据至关重要,Rtsne 包的出现,为我们提供了丰富多彩的方式来呈现和分析数据。对于高维数据的降维有许多种方法,我们今天介绍其中的一种:tSNE(t-Distributed Stochastic Neighbor Embedding,t分布随机近邻嵌入),tSNE是目前效果最好的数据降维和可视化方法。我们可以更加有效地降维与可视化,从而更好地理解和解释数据。
练了十年生信的小果要凭本事吃饭了,要是您有自己做不了的生信分析,可以联系我。
本次介绍的R包需要较多的硬件资源,在服务器可以更加流畅运行,同学们如果没有自己的服务器欢迎联系我们使用服务器租赁~
tSNE与PCA
tSNE与PCA的区别主要在于tSNA是一种非线性降维方法,而PCA是一种线性降维方法。PCA有可能导致拥挤现象,即在原本的高维空间中明显是多簇的数据点,有可能会被降维至一处,几类数据边界不明显,混杂在一起,没有被很好地分开,显得十分“拥挤”。
我们可以想象一块面包,表面铺了一层芝麻,中心是豆沙馅。线性降维方法很好理解,就是以某种方向将这个面包拍扁,让豆沙和芝麻在平面上分开来。三维面包的豆沙馅和芝麻很显然是分开的,但是如果你垂直把他拍扁,那么表层的芝麻和内里的豆沙馅无可避免地会混在一起。当然实际情况的PCA没有这么蠢,它可能会把面包竖起来再拍扁,这样就可以想象降维后的面包芝麻聚集在一条线上,而豆沙馅在旁边的一团。
但是如果这个面包师喜欢往面团里加花生碎怎么办?成品的面包,豆沙馅料,花生碎的分布围绕着豆沙馅,表面是芝麻。花生碎在三维面包中与芝麻、豆沙都没有交集,很容易区分,但也很容易想到,无论你如何将这个面包直接拍扁,都至少有两团料会混在一起。
而 t-SNE 更加注重保留原始数据的局部特征,就可以很好地规避拥挤现象。简单来说,t-SNE 中主要是将“距离的远近关系”转化为一个概率分布,每一个概率分布就对应着一个“样本间距离远近”的关系。
Rtsne包介绍
Rtsne包是一个R语言扩展包,专注于tSNE分析。
Rtsne包安装
需要R语言版本为4.4,在控制台中输入以下命令:
install.packages(“Rtsne”)
查看是否安装成功
packageVersion(“Rtsne”) # 查看Rtsne版本
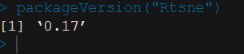
显示为0.17版本及以上,则表示已经安装了Rtsne包。
使用Rtsne包进行降维分析与可视化
为了演示Rtsne包的用法,本文中我们将使用随机生成的树文件和模拟的关联数据集,使用的模拟数据集都包含树的那一列分类标签。
载入包:
library(Rtsne) # 载入Rtsne包
另外还需要R语言自带的base包,不需要下载,直接加载即可:
library(base) # 载入base包
载入数据:
我们用到的是R自带的鸢尾花(iris)数据集:
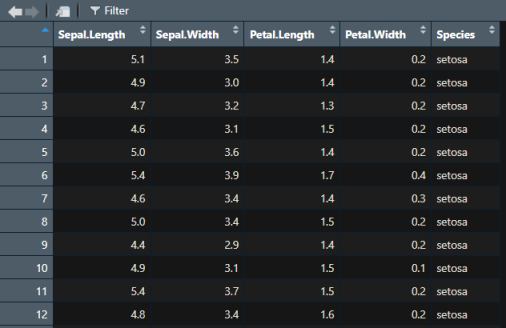
按每行来看,它是一个四维的数据,分别以Sepal.Length(萼片长度)、Sepal.Width(萼片宽度)、Petal.Length(花瓣长度)、Petal.Width(花瓣宽度)为坐标轴,写成坐标点的形式就是(萼片长度, 萼片宽度, 花瓣长度, 花瓣宽度),是一个四维的数据。
如果用PCA对其进行降维:
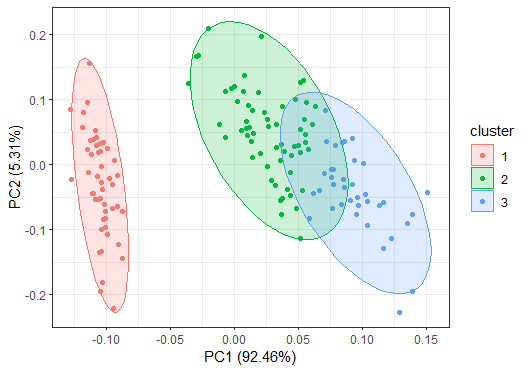
图中可以看出,红点的1类和绿点的2类、蓝点的3类分得比较开,但是蓝点和绿点是有一部分混杂在一起的,没有很好地分开。而且这是唯一的,只要你用PCA就只能降维成这个样子。
现在我们用tSNE对其进行降维:
library(Rtsne)
iris_unique <- unique(iris) # 去除重复值
tsne_out <- Rtsne(as.matrix(iris_unique[,1:4])) # 运行tSNE
plot(tsne_out$Y,col=iris_unique$Species,pch=19)# 结果绘图
结果如下图:
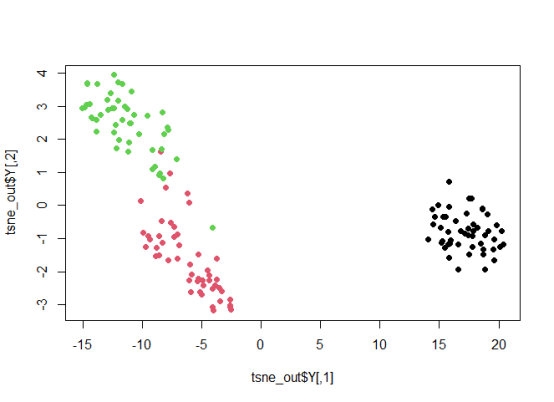
tSNE的名字里面有个“随机”,所以每次输出的图片都不一样。这些图片里面有比较好的,也有比较差的,可以通过设置随机数种子来获得比较满意的图片,同时保证复现性。一般来说我们不知道哪个随机数好哪个差,所以我们可以通过死循环来遍历。
a <- 1 # 随机数种子初始值
while(T){
set.seed(a) # 设置随机数种子,保证复现性
print(a) # 输出随机数种子
tsne_out <- Rtsne(as.matrix(iris_unique[,1:4])) # 运行tSNE
plot(tsne_out$Y,col=iris_unique$Species,pch=19) # 结果绘图
a <- a + 1 # 随机数种子+1
Sys.sleep(3) # 睡眠3秒
}
在这个死循环中,我设置了随机数种子的初始值为1,然后进入死循环。循环体中,首先输出此次的随机数种子,然后以这个随机数种子进行一次tSNE分析与绘图,绘图完成后随机数种子+1,睡眠3秒让你可以看一看这个图好不好,然后进行下一次循环。
要注意的是,我并没有设置循环的退出条件,如果你不手动停止的话,这个循环永远不会结束,每隔3秒出一张图。如果你想停止了,就点控制台右上角的小红STOP“Interrupt R(打断R)”手动停止循环。一定要记得停止!否则这个死循环会永远运行下去!!
我进行了100次循环,发现随机数种子设为44时的图片我最满意,于是记下来,在第一次出现的代码中插入一句就可以了:
iris_unique <- unique(iris) # 去除重复值
set.seed(44) # 100以内我认为最好的随机数种子
tsne_out <- Rtsne(as.matrix(iris_unique[,1:4])) # 运行tSNE
plot(tsne_out$Y,col=iris_unique$Species,pch=19)# 结果绘图
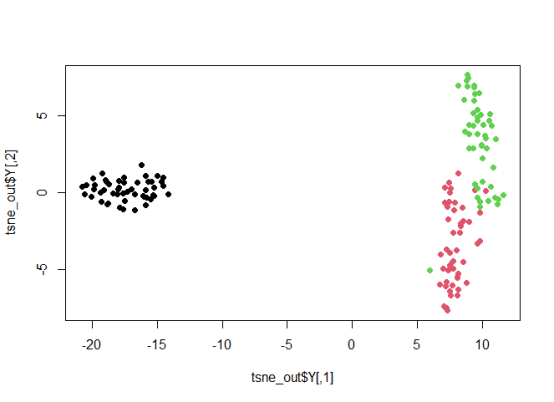
由图可见,iris数据集中始终有两组数据难分难解,但已经比PCA好了不少。如果循环到以后,可能出现更好的图片,这就要用户自己的考量了。
至此,我们绘制出了使用tSNE降维的鸢尾花数据集,图所能展现的信息更加多元化,数据之间的对应关系更加明确,读者可以轻松获取降维后的信息。
以上就是对于Rtsne包的全部介绍了,在本文中,我们详细介绍了如何使用 Rtsne 包来进行降维与可视化。同学们可以通过学习和应用 Rtsne 包,掌握如何在 R 语言环境下进行系统发育树的可视化,并且将树形结构与其他数据信息相结合,从而更好地理解和解释数据。这不仅有助于提升数据分析的效率,还能够让研究结果更具可解释性和可视化效果。小果希望同学们继续深入学习和探索Rtsne 包的相关知识,不断尝试新的工具和技术,提升自己的数据分析能力和科研水平。
同学们如果觉得自己写代码麻烦,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表。立即访问云生信(http://www.biocloudservice.com/home.html),开启便捷的生信之旅!