大家好~我是小果!在这个信息爆炸的时代,基因表达数据成为了解锁生命科学秘密的关键。作为生信领域的探索者,我们深知基因表达数据它不仅关系到疾病的诊断与治疗,还涉及到生物进化和个体差异的深层理解。今天,小果将带领大家一起探索如何将这些复杂的数据转化为有价值的信息。从数据开始通过精心设计的预处理步骤,筛选出关键信息,去除那些可能干扰的噪声。再运用统计学的强大工具,挖掘数据中的模式和关联。最后,通过直观的可视化,将这些复杂的数据转化为易于理解的图形。在这个过程中,小果将介绍一系列实用的R软件包,它们将是我们探索之旅中的得力助手。Limma包、stats包、ggplot2包、ggrepel包以及pheatmap包,每一个都有其独特的功能和优势,它们将帮助我们从不同角度分析和展示数据。小果会通过实际的代码示例,让大家看到每一步是如何操作的。现在就和小果一起揭开基因表达数据的神秘面纱吧!同时呢,十年的生信之路,小果已经练就了一身扎实的本领,现在准备用这身本事为小伙伴们服务啦!如果你在生信分析上遇到了难题,那就来找小果吧!小果会用自己的专业知识和技能,为你解决困扰,助你一臂之力。期待你的联系哦~
一、 用到的包简要介绍
- Limma包
Limma包是专为微阵列数据和RNA-seq数据分析而设计的R软件包,不仅支持cDNA芯片的RAW数据输入和预处理(包括归一化等)功能,还拥有独特的“线性”算法,特别适用于多因素实验。
Limma包在处理大规模数能够很好地控制假阳性率。其分析过程包括ANOVA分析和线性回归等,对于单通道和双通道数据都能进行分析,甚至包括定量PCR和RNA-seq数据。
- 小果提示:在使用Limma包时,通常要求输入的表达矩阵是经过log2处理之后的。
- stats包
stats是R语言中一个非常基础的包, stats包提供了多种统计分析函数,包括描述性统计(如均值、中位数等)、概率计算、假设检验、线性回归等。除了统计分析外,stats包还提供了多种统计图形的绘制功能,如直方图、散点图、箱线图等。
- ggplot2包和ggrepel包
ggrepel包主要用于解决在数据可视化过程中标签重叠的问题。ggrepel包提供了一系列函数和工具,用于自动调整标签的位置,从而提高图形的可读性和吸引力。ggrepel包是基于ggplot2的插件,因此与ggplot2具有良好的兼容性。
- pheatmap包
pheatmap包是R语言中的一个用于绘制聚类热图的强大工具。这个包提供了一系列直观的函数和参数,使得我们能够轻松地自定义热图的外观和布局。
二、 代码实操
废话不多说,小果直接带小伙伴们来实操代码,进行基因表达数据处理的全面示例!本次介绍的R包操作会占用内存较大,小果建议使用服务器,欢迎小伙伴们联系小果租赁性价比居高的服务器!
- 数据生成:小果首先使用set.seed确保结果的可重复性。然后,创建一个数据框data,包含100个基因(Gene),每个基因的Fold Change和Pvalue是随机生成的,条件(condition)分为控制组(control)和处理组(treatment),每组50个基因。
set.seed(123)
data <- data.frame(
Gene = paste(“Gene”, 1:100),
FoldChange = runif(100, min = -3, max = 3),
Pvalue = runif(100, min = 0, max = 1),
condition = factor(rep(c(“control”, “treatment”), each = 50))
)
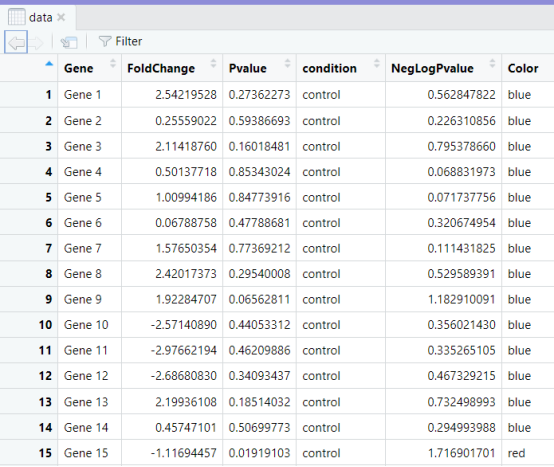
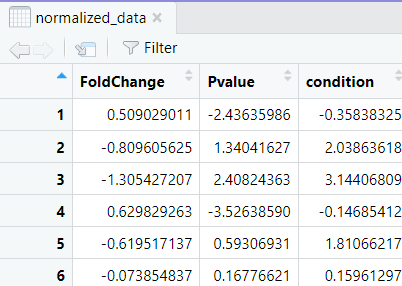
- 数据预处理:小果接着使用了limma包中的函数将非数值型数据转换为数值型。对数据进行标准化处理,首先减去每行的均值,然后除以每列的标准差。
library(limma)
data[, -1] <- sapply(data[, -1], as.numeric)
normalized_data <- sweep(data[, -1], 1, rowMeans(data[, -1], na.rm = TRUE), “-“)
normalized_data <- sweep(normalized_data, 2, apply(data[, -1], 2, sd, na.rm = TRUE), “/”)
head(normalized_data)

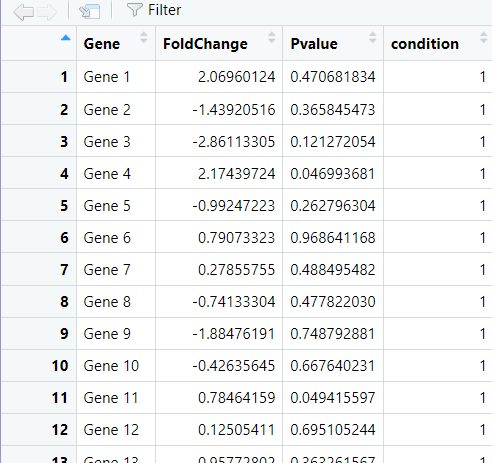
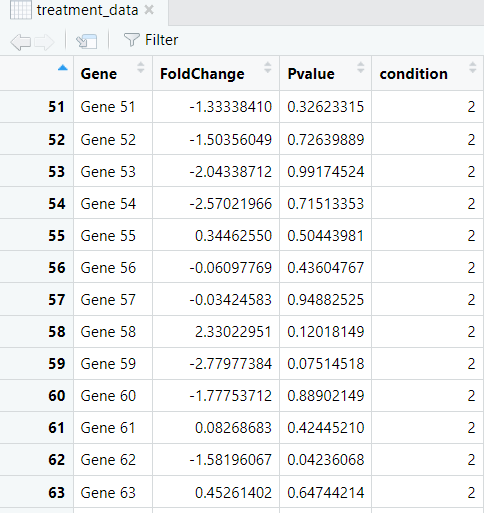
- 数据分离:小果根据相关条件将数据分为了控制组和处理组。
control_data <- data[data$condition ==1, ]
treatment_data <- data[data$condition == 2, ]
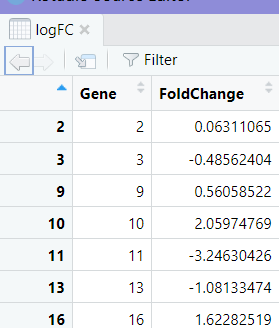
- 计算对数变化倍数(logFC):对控制组和处理组的Fold Change进行对数变化倍数的计算,并且去除NA值。
logFC <- data.frame(Gene=1:50,
FoldChange = log2(treatment_data$FoldChange / control_data$FoldChange))
logFC <- na.omit(logFC)
head(logFC)
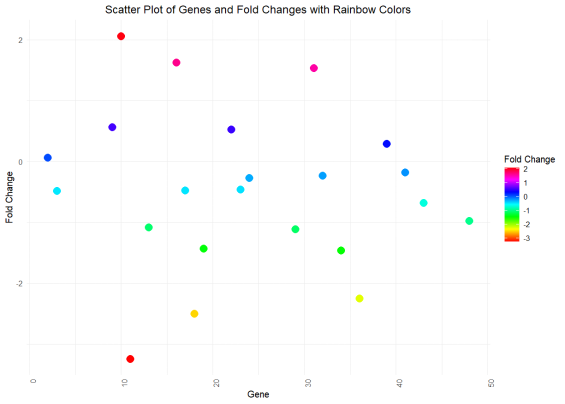
- 可视化绘图:使用ggplot2绘制的散点图展示了基因(Gene)与对数变化倍数(Fold Change)之间的关系。基因名称在x轴上,对数变化倍数在y轴上。点的大小设置为4,颜色根据Fold Change的值变化,使用彩虹色渐变,这样可以直观地看出哪些基因表达变化较大。然后绘制基因和Fold Change的条形图。
ggplot(logFC, aes(x = Gene, y = FoldChange, color = FoldChange)) +
geom_point(size = 4) +
scale_color_gradientn(colors = rainbow(100), limit = range(logFC$FoldChange), na.value = “grey50”) +
theme_minimal() +
labs(title = “Scatter Plot of Genes and Fold Changes with Rainbow Colors”,
x = “Gene”,
y = “Fold Change”,
color = “Fold Change”) +
theme(axis.text.x = element_text(angle = 90, hjust = 1), plot.title = element_text(hjust = 0.5))
ggplot(logFC, aes(x = reorder(Gene, FoldChange), y = FoldChange)) +
geom_bar(stat = “identity”, fill = “skyblue”, color = “black”, width = 0.5) +
theme_minimal() +
labs(title = “Bar Plot of Genes and Fold Changes”,
x = “Gene”,
y = “Fold Change”) +
theme(axis.text.x = element_text(angle = 45, hjust = 1, vjust = 1, size = 8),
axis.title.x = element_blank(),
axis.title.y = element_text(size = 12),
plot.title = element_text(hjust = 0.5)) +
coord_flip()

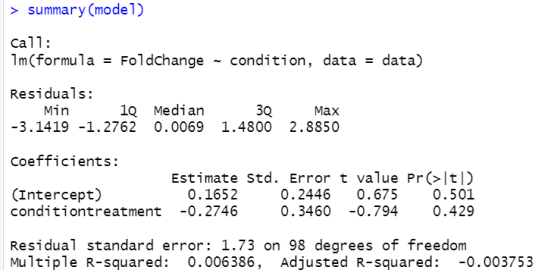
- 线性模型分析:使用limma包中的函数创建设计矩阵。拟合线性模型,分析不同条件对基因表达的影响。
data$condition <- factor(data$condition, levels = c(“control”, “treatment”))
design <- model.matrix(~condition, data = data)
print(design)
model <- lm(FoldChange ~ condition, data = data)
summary(model)
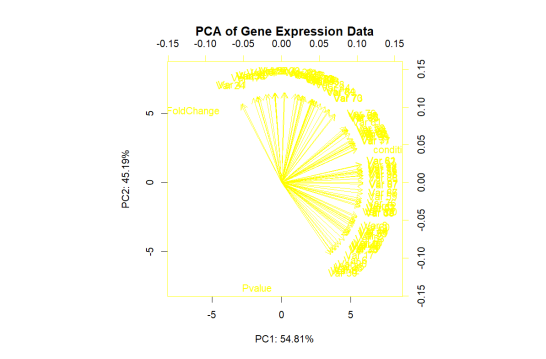
- 主成分分析(PCA):对标准化后的数据进行PCA分析,并绘制双主成分得分图。PCA图展示了基因表达数据在两个主成分方向上的分布情况。第一主成分(PC1)和第二主成分(PC2)分别解释了数据变化的百分比,这些成分可以用于区分不同的条件或群体。
library(stats)
pca <- prcomp(normalized_data, scale. = TRUE)
summary(pca)

biplot(pca$x[, 1:2], pca$rotation[, 1:2],
main = “PCA of Gene Expression Data”,
xlab = “PC1: 54.81%”,
ylab = “PC2: 45.19%”,
col = ifelse(pca$x[, 1] > 0, “green”, “yellow”))
- 火山图:绘制火山图用于基因表达数据分析,以Fold Change为x轴,负对数P值(-Log10 P-value)为y轴。显著性阈值(如P < 0.05)通常用来区分显著和非显著的基因。颜色区分显著性,例如红色可能表示P值小于0.05,而蓝色表示P值大于或等于0.05。火山图可以帮助识别表达差异显著的基因。
library(ggplot2)
library(ggrepel)
data$NegLogPvalue <- -log10(data$Pvalue)
data$Color <- ifelse(data$Pvalue < 0.05, “red”, “blue”)
ggplot(data, aes(x = FoldChange, y = NegLogPvalue, color = Color)) +
geom_point(alpha = 0.8,size=5) +
scale_color_manual(values = c(“red”=”red” ,”blue”=”blue”)) +
theme_minimal() +
labs(title = “Volcano Plot”, x = “Fold Change”, y = “-Log10 P-value”) +
theme(legend.title = element_blank()) +
geom_text(data = data[data$Pvalue < 0.05, ],
aes(label = Gene),
vjust = -1, hjust = 1, size = 3, color = “black”)

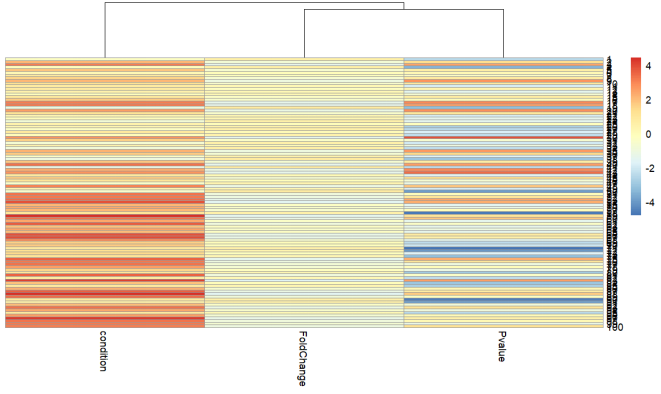
- 热图:使用pheatmap包绘制基因表达数据的热图,根据条件进行颜色注释。热图展示了基因表达数据的矩阵视图,行代表基因,列代表样本。
颜色的深浅表示表达水平的高低,通常用于观察基因表达模式和样本之间的相似性。
if (!requireNamespace(“pheatmap”, quietly = TRUE)) {
install.packages(“pheatmap”)
}
library(pheatmap)
pheatmap(normalized_data,
cluster_rows = FALSE,
show_rownames = TRUE,
annotation_col = data$Condition)
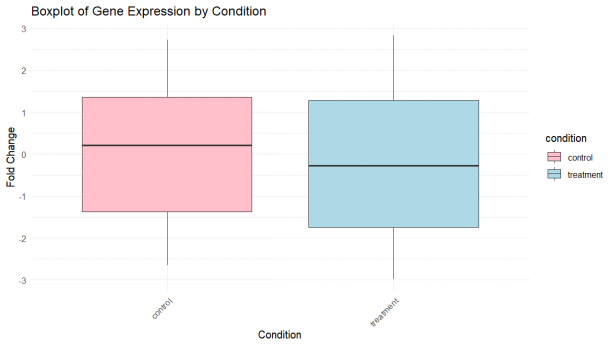
- 箱线图:绘制箱线图,展示不同条件下基因表达的分布情况。箱线图展示了不同条件下基因表达的分布情况,包括中位数、四分位数以及异常值。颜色区分不同的条件,例如“control”和“treatment”,有助于比较两组之间的表达差异。
ggplot(data, aes(x = condition, y = FoldChange, fill = condition)) +
geom_boxplot() +
scale_fill_manual(values = c(“control” = “pink”, “treatment” = “lightblue”)) +
theme_minimal() +
labs(title = “Boxplot of Gene Expression by Condition”,
x = “Condition”,
y = “Fold Change”) +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
三、 文章小结
本期小果和小伙伴们一起学习了一个完整的基因表达数据分析流程,包括数据生成、预处理、统计建模、以及多种可视化技术。我们首先创建了一个模拟数据集,随后通过标准化处理使数据满足分析的前提条件。接着,代码分离出控制组和处理组的数据,并计算了对数变化倍数(logFC)。在可视化部分,使用了ggplot2包来绘制散点图和火山图,以及pheatmap包来生成热图,这些图表直观地展示了基因表达的差异和统计显著性。此外,还进行了主成分分析(PCA)以探索数据的内在结构,并通过箱线图比较了不同条件下的基因表达分布。让我们继续探索生信的无限可能,共同开启更多科研新篇章!最后如果各位小伙伴们觉得自己运行代码太麻烦,欢迎用我们的云生信小工具,只要输入合适的数据就可以直接出想要的图哦,附云生信链接(http://www.biocloudservice.com/home.html)。