小伙伴们,是否曾为复杂的基因组注释数据而头疼不已?别担心,小果今天给大家带来了一位生信界的救星——GenomeInfoDb R包!这个强大的R语言工具包可是专门用于存储和查询基因组注释信息的,它能助你一臂之力,让生物信息学分析变得轻松又高效!无论是初学者还是资深玩家,GenomeInfoDb R包都能为你提供强大的支持。它能帮助你管理和整合各种基因组注释数据,让你的分析工作更加得心应手。而且呢,小果在这里要告诉大家一个好消息!如果你有生信分析方面的难题,不妨联系小果哦!练了十年生信的小果可是有着丰富的经验和实力,定能为你提供专业的帮助和解答。希望本期内容能够对你有所帮助,让生信分析不再是你的绊脚石!一起加油,开启生物信息学分析的新篇章吧!
(一) GenomeInfoDb R包基本介绍
GenomeInfoDb包是一个用于存储和查询基因组注释信息的R语言工具包。它可以帮助管理和整合各种基因组注释数据,包括基因名称、染色体位置、转录本信息等。通过GenomeInfoDb包,小伙伴们可以创建自定义的基因组注释数据库,以便在后续的生物信息学分析中使用。
GenomeInfoDb包不仅功能强大,而且非常灵活。它能够支持多种基因组注释数据的导入,无论是基因名称、染色体位置还是转录本信息,都能轻松应对。更重要的是,它允许小伙伴们根据自己的需求进行定制,创建属于自己的基因组注释数据库。这样,在后续的生物信息学分析中,我们就可以轻松地使用这些注释数据,提高分析的准确性和效率。
那么,GenomeInfoDb包具体在哪些领域发挥着重要作用呢?
- 在基因表达分析中,我们可以利用GenomeInfoDb包来查询和整合不同基因的表达数据,从而更深入地了解基因的功能和调控机制。
- 在基因变异注释方面,GenomeInfoDb包可以帮助我们快速定位和分析基因组中的变异位点,揭示变异对基因功能的影响。
- 在代谢通路分析中,通过利用GenomeInfoDb包,我们可以更好地理解代谢途径中各个基因的作用和相互关系。
(二) GenomeInfoDb R包代码实操
那么废话不多说,小果直接带小伙伴们上代码实操!
小果这里将以GenomeInfoDb R包在不同方面的应用进行举例~小伙伴们要是觉得自己下载R包操作太麻烦,可以使用相关服务器哦~欢迎联系小果租赁性价比居高的服务器!小果随时恭候!
- 基因表达分析
- 安装和加载所需的R包
library(GenomeInfoDb)
library(GenomicFeatures)
library(DESeq2)
- 从GenomeInfoDb包中获取基因信息:使用makeTxDbFromUCSC函数从UCSC数据库中获取人类基因组注释信息,并将其存储在txdb对象中
txdb <- makeTxDbFromUCSC(“hg19”)
- 从GenomicFeatures包中获取基因组注释信息
- 使用genes函数从txdb中提取基因信息,并将其存储在genes对象中
genes <- genes(txdb)
exons <- exonsBy(txdb, by=”gene”)
- 基因表达数据,这里使用DESeq2包进行基因表达分析
set.seed(123)
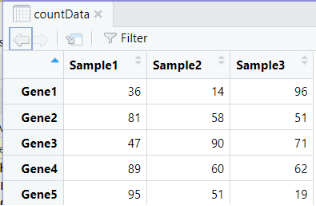
countData <- matrix(data = round(runif(15, min = 10, max = 100)), nrow = 5, ncol = 3)
colnames(countData) <- c(“Sample1”, “Sample2”, “Sample3”)
rownames(countData) <- c(“Gene1”, “Gene2”, “Gene3”, “Gene4”, “Gene5”)
- 创建colData:创建实验信息colData,包括样本名称和条件信息(这里假设有两种条件:Control和Treatment)
sample_names <- c(“Sample1”, “Sample2”, “Sample3”)
conditions <- c(“Control”, “Treatment”, “Control”)
colData<-data.frame(sampleName=sample_names,condition = factor(conditions))
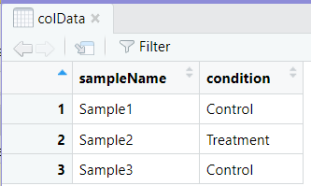
countData是基因表达计数数据,colData是实验信息(小伙伴们可以根据自己的实际需求代换数据集!)
- 使用DESeqDataSetFromMatrix函数创建DESeq2数据集对象dds,并指定差异表达分析的设计矩阵
dds <- DESeqDataSetFromMatrix(countData, colData, design=~condition)
- 使用DESeq函数对dds进行差异表达分析;results函数提取差异表达分析的结果,并将结果存储在res对象中
dds <- DESeq(dds)
res <- results(dds)
- 输出差异表达基因:提取差异表达基因中的前10个基因名topGenes,并根据基因名在genes对象中提取相应的基因信息topGenesInfo
topGenes <- head(rownames(res), 10)
topGenesInfo <- genes[rowRanges(dds)$gene_id %in% topGenes,]
print(topGenesInfo)

- 基因变异注释
- 相关R包的安装
if (!requireNamespace(“BiocManager”, quietly = TRUE))
install.pack.packages(“BiocManager”)
BiocManager::install(“GenomeInfoDb”)
library(GenomeInfoDb)
library(GenomicRanges)
library(GenomicFeatures)
library(DESeq2)
- 查询基因组注释数据(小果这里就以TP53举例了,小伙伴们可根据自己实际需求进行修改):查询特定基因(TP53)的注释信息、转录本信息和外显子信息,并存储在query_gene、query_transcript和query_exon对象中
transcripts <- transcripts(txdb)
exons <- exons(txdb)
query_gene <- genes[genes$gene_name == “TP53”, ]
query_transcript <- transcripts[transcripts$transcript_name == “NM_000546.5”, ]
query_exon <- exons[exons$exon_id == “ENSE00003554982”, ]
- 获取基因组坐标信息:使用mapToGenome函数获取特定基因(TP53)的基因组坐标信息,并存储在gene_coords、transcript_coords和exon_coords对象中
gene_coords <- mapToGenome(“TP53”, txdb)
transcript_coords <- mapToGenome(“NM_000546.5”, txdb)
exon_coords <- mapToGenome(“ENSE00003554982”, txdb)
- 获取基因组序列信息:使用getSeq函数获取特定基因(TP53)的基因组序列信息,并存储在gene_seq、transcript_seq和exon_seq对象中
gene_seq <- getSeq(txdb, “TP53”)
transcript_seq <- getSeq(txdb, “NM_000546.5”)
exon_seq <- getSeq(txdb, “ENSE00003554982”)
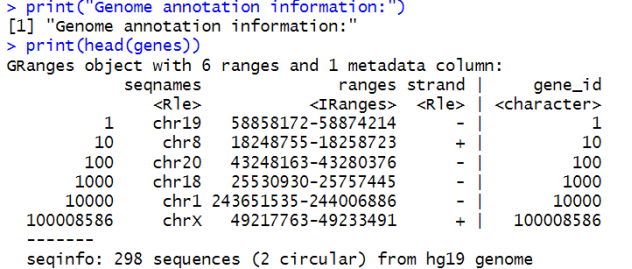
- 输出相关结果:包括基因组注释信息、查询结果、基因组坐标和基因组序列信息
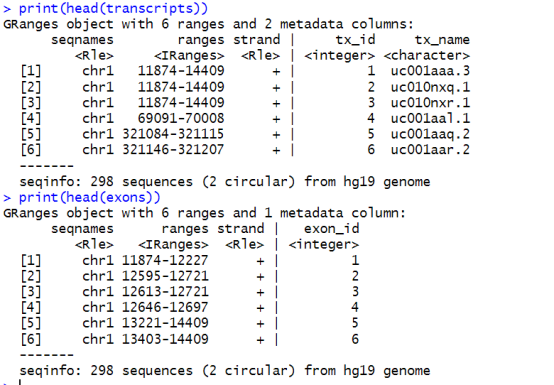
print(“Genome annotation information:”)
print(head(genes))
print(head(transcripts))
print(head(exons))
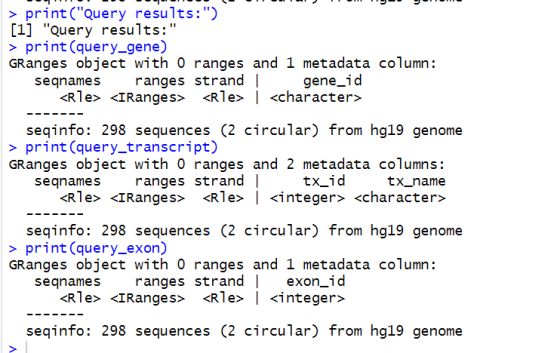
print(“Query results:”)
print(query_gene)
print(query_transcript)
print(query_exon)
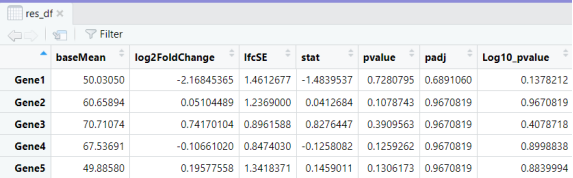
- 相关可视化
- 将 res 对象转换为数据框 res_df,并添加两个新列 pvalue 和 Log10_pvalue,绘制了五个图形进行可视化:基因表达水平的箱线图、平均表达水平与对数倍数变化的散点图(MA图)、箱线图等。其中,scale_color_manual 和 scale_fill_manual 用于自定义颜色,labs 用于设置图形的标题和轴标签。
library(ggplot2)
res$pvalue <- 10^-abs(res$pvalue)
res$Log10_pvalue <- -log10(res$pvalue)
res_df <- as.data.frame(res)
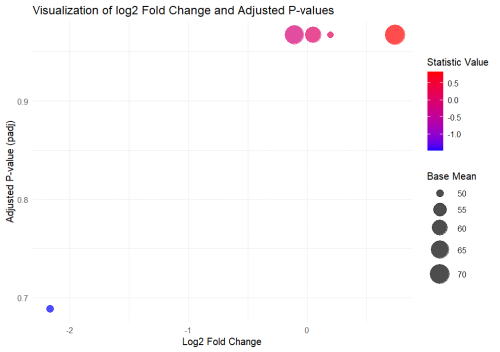
ggplot(res_df, aes(x = log2FoldChange, y = padj, size = baseMean, color = stat)) +
geom_point(alpha = 0.7) +
scale_size_continuous(range = c(3, 10)) +
scale_color_gradient(low = “blue”, high = “red”) +
theme_minimal() +
labs(title = “Visualization of log2 Fold Change and Adjusted P-values”,
x = “Log2 Fold Change”,
y = “Adjusted P-value (padj)”,
size = “Base Mean”,
color = “Statistic Value”)
res_df$significant <- ifelse(res_df$padj < 1.0, “Significant”, “Not Significant”)
ggplot(res_df, aes(x = “”, y = baseMean, fill = significant)) +
geom_boxplot() +
theme_minimal() +
scale_fill_manual(values = c(“pink”)) +
labs(title = “Boxplot of Gene Expression”, x = “”, y = “Base Mean”, fill = “Significance”)
ggplot(res_df, aes(x = “”, y = log2FoldChange, fill = factor(padj < 1.0))) +
geom_violin(trim = FALSE, scale = “count”) +
theme_minimal() +
labs(title = “Density Plot of Log2 Fold Change”, fill = “Significant”)
ggplot(res_df, aes(x = baseMean, y = log2FoldChange)) +
geom_point(aes(color = padj < 1.0), alpha = 0.8) +
scale_color_manual(values = c(“green”)) +
theme_minimal() +
labs(title = “MA Plot”, x = “Average Expression Level (baseMean)”, y = “Log2 Fold Change”)
ggplot(res_df, aes(x = factor(padj < 1.0), y = log2FoldChange, fill = “factor(padj <1.0)”)) +
geom_bar(stat = “summary”, fun = “mean”, position = position_dodge()) +
theme_minimal() +
labs(title = “Mean Log2 Fold Change by Significance”)
(三) 文章小结
本期,小果为小伙伴们分享了GenomeInfoDb 包的应用实例,它在生物信息学领域中发挥着举足轻重的作用。作为一套高效且灵活的工具集为我们提供了管理和整合基因组注释数据的便捷途径,显著提升了分析的准确性和效率。它提供了强大的数据结构和函数,使我们能够轻松地处理复杂的基因组注释数据。它不仅支持多种格式的数据输入,而且能够将这些数据整合成一个统一的结构,方便后续的分析操作。这对于我们来说,能够更加专注于生物学问题的研究,而无需花费大量时间在数据格式的转换和整合上。
其次,GenomeInfoDb 包让我们可以方便地查询基因的位置、染色体信息、转录本结构等关键信息,从而更加全面地了解基因组的结构和功能。同时该包还支持与其他生物信息学工具的集成,能够轻松地将基因组注释数据与其他类型的数据进行联合分析,进一步挖掘生物学信息。并且它提供了严格的数据校验和质量控制机制,确保所使用的基因组注释数据的准确性和完整性。这对于生物信息学分析来说至关重要,因为不准确的数据可能导致误导性的分析结果。
总之,GenomeInfoDb包是生物信息学领域的一款神器,它帮助我们轻松管理和整合基因组注释数据,提高了分析的准确性和效率。如果你还在为复杂的基因组注释数据而头疼不已,不妨试试GenomeInfoDb包吧!相信它一定会成为你生物信息学分析中的得力助手。如果各位觉得自己运行代码太麻烦,欢迎用我们的云生信小工具,只要输入合适的数据就可以直接出想要的图呢,附云生信链接(http://www.biocloudservice.com/home.html)