
同学们,大家好!今天小果要想大家介绍一个非常强大的多序列比对工具——muscle包。作为一款高效的多序列比对工具,muscle包在生物信息学研究中发挥着重要的作用。它能够对DNA、RNA、蛋白质序列进行高效且精确的比对,是我们在序列分析中不可或缺的助手。
muscle包不仅操作简单,而且功能强大,可以处理大量的序列数据,进行复杂的比对任务。更重要的是,muscle包与ggmsa包的结合,使得我们可以轻松地将比对结果进行可视化展示,让复杂的生物数据一目了然。任何生信分析对生信高手小果来说都不是难题,要是同学们有自己做不了的生信分析,欢迎随时联系我!!!
接下来,我们将一步步学习muscle包的安装方法、如何载入示例数据、进行多序列比对以及利用ggmsa包进行结果的可视化展示。通过实例演示,我们将对31种哺乳动物的MAX基因序列进行比对,揭示其在不同物种中的保守性和变异情况。让我们一起踏上这段学习之旅,深入探索muscle包的强大功能,掌握多序列比对的技巧,推动自己的研究进展。准备好了吗?让我们开始吧!
本次介绍的R包需要较多的硬件资源,在服务器可以更加流畅运行,同学们如果没有自己的服务器欢迎联系我们进行服务器租赁~
muscle包介绍
muscle是一个提供了对Muscle多序列比对算法的接口和功能的R语言包。它允许用户在R环境中轻松地进行多序列比对的分析和处理。muscle包提供了一系列函数,可以方便地调用Muscle算法执行多序列比对,并将结果以适合R语言环境的数据结构返回,使用户能够方便地进行后续的数据分析和可视化。该包还提供了丰富的参数选项,允许用户根据需要调整比对的精度和速度。通过muscle包,用户可以在R中灵活地进行多序列比对的研究和分析,为生物信息学领域的研究提供了便利的工具。
muscle包的安装
需要R语言版本为4.4,在控制台中输入以下命令:
if (!require(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager “)
BiocManager::install(“muscle”) # 在BiocManager环境下安装muscle
查看是否安装成功
packageVersion(“muscle”) # 查看muscle版本

显示为3.46.0版本,则表示已经成功安装了muscle包。
除此之外,后续示例还需要使用ggmsa包,我们可以提前安装,安装命令如下:
BiocManager::install(“ggmsa”) # 在BiocManager环境下安装ggmsa
packageVersion(“ggmsa”) # 查看ggmsa版本

显示为1.10.0版本,则表示已经成功安装了muscle包。
至此,我们已经安装了所有需要用到的R包,接下来我们就正式开始对muscle包的介绍,干货满满,同学们要认真听讲哦。
使用muscle包进行多序列比对并可视化
包和数据的载入:
首先我们需要载入muscle包,ggmsa包在后续进行结果可视化的部分需要用到,我们可以提前载入。除此之外本篇采用的示例数据为31种哺乳动物的MAX基因序列,该示例数据整合在muscle包内,我们可以直接调用使用,相关命令如下:
# 载入muscle包
library(muscle)
# 载入ggmsa包
library(ggmsa)
# 输出umax对象内容
print(umax)
# 显示umax对象类别
class(umax)

显示如上图,可知umax对象类型为DNAStringSet,共包含31条MAX基因的序列信息,这些基因的大致长度在400-500bp之间,我们可以打印序列的名称,查看其包含的物种,命令如下:
# 打印序列名称
names(umax)
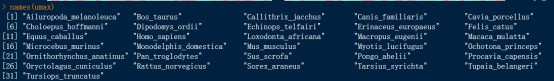
显示如上图,可知umax数据集涵盖的物种丰富,包括大熊猫(Ailuropoda_melanoleuca)、牛(Bos_taurus )、人类(Homo_sapiens)等在内的31个物种的MAX基因。
使用muscle包进行多序列比较
我们可以直接使用muscle包种的muscle()函数,进行多序列比较,muscle()函数的参数丰富,可以满足多种序列比对的需要,如命令
muscle(sequences,verbose= FALSE,gap_open= -400,gap_extend=-80)其中:
verbose: 一个逻辑值,控制是否输出额外的信息来显示比对的进度和细节。默认为FALSE,表示不输出额外信息。
gap_open: 设置开放缺失值惩罚的值。这是一个非负数,用于惩罚在序列比对中打开一个缺失值。默认值为-400。
gap_extend: 设置延长缺失值惩罚的值。这是一个非负数,用于惩罚在序列比对中延长一个已经存在的缺失值。默认值为-80。
为了演示简单易懂,我们使用muscle()函数均采用默认参数,相关命令如下:
#使用muscle()函数将umax基因簇中的基因进行多序列比对
aln <- muscle(umax, verbose= FALSE, gap_open= -400, gap_extend=-80)
#打印比对结果
aln

结果显示如上图,我们可以看到MAX基因的3‘端和5’端均具有大量的保守位点,在不同物种中均具有相同的碱基序列,为了更好的展示结果,我们可以使用ggmsa包来绘制DNA多序列比对图,相关命令如下:
# 调用ggmsa函数来创建多序列比对的图形化表示
# color: 设置序列的颜色方案,这里使用了Zappo_AA颜色方案,font: 设置字体,这里使用了DroidSansMono字体,char_width: 设置字符宽度,控制序列的显示宽度,seq_name: 控制是否显示序列名称
ggmsa(aln, color = “Zappo_AA”, font = “DroidSansMono”, char_width = 0.5, seq_name = TRUE ) +
# 添加序列Logo图
geom_seqlogo(color = “Zappo_AA”) +
# 添加多序列比对的标尺
geom_msaBar()

结果如上图,可知MAX基因在31种哺乳动物中整体差异较小,只有个别位点发生突变,这表明MAX基因在哺乳动物中可能具有高度保守性,MAX基因编码一种叫做MAX的蛋白,它是一种转录因子,参与调控细胞生长和分化等重要生物学过程,MAX基因的保守性意味着MAX蛋白在不同哺乳动物中的功能需求类似,因此基因序列保持相对稳定。个别位点的突变可能是由于个体间的遗传变异或环境压力等因素导致的,这些突变可能对MAX基因的功能产生一定影响,但由于整体上保守性较高,这些影响可能并不会导致基因功能的显著改变。同学们可以结合自己课题的序列信息,对多序列比对结果进行解释说明。
以上就是对于muscle包的全部介绍了,在本文中,我们详细介绍了muscle包在R语言环境下的安装和使用方法,并通过实例演示了如何利用muscle包对31种哺乳动物的MAX基因序列进行多序列比对。通过本文的介绍,同学们不仅了解了muscle包的基本使用方法,还掌握了如何结合ggmsa包进行结果的可视化展示。这些技能对于分析和解释多序列比对结果具有重要意义。希望同学们在今后的学习和研究中,能够灵活应用muscle包及相关工具,深入分析生物序列数据,推动自己的研究进展。继续努力学习muscle包,相信你们会发现更多的应用场景和潜在的研究价值。
同学们如果觉得自己写代码麻烦,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表。立即访问云生信(http://www.biocloudservice.com/home.html),开启便捷的生信之旅!