大家好啊,在公共数据库中我们还经常会遇到转录组数据,转录组数据是无法用R包下载的,因为转录组数据的Series Matrix File(s)没有基因表达量数据,只有临床数据,所以每次处理转录组数据我们都要在Supplementary file下载文件然后处理,但是很不巧的是很少有作者会上传他们整合好的数据,大部分作者都会上传未整合的数据,这时候我们需要从头开始整合数据。
从头开始整合数据已经很麻烦了,有的作者甚至上传的不是未整合的表达量数据,比如GSE104140和GSE58150,他们上传bw或wig文件,令人窒息,虽然有文献提到可以用bw文件来存储RNA-seq数据,但是小花没有找到如何才能将bw文件转为基因表达量文件。所以这时候我们需要下载最原始的fastq文件来从头开始处理,听到这里也许你已经开始头大了,但是不要慌,小花手把手教你如何从数据下载到数据处理到得到结果,带你体验飞一般的感觉。要是您有自己做不了的生信分析,可以联系我。要进行下面的操作您还需要一个服务器,欢迎联系小花租赁高性价比的服务器哦。
然后我们开始准备下载数据吧,一般的方法就是使用sra-toolkit的prefetch来下载数据。以GSE58150为例,首先在NCBI的GEO数据库搜索GSE58150,然后在最下面找到SRA Run Selector
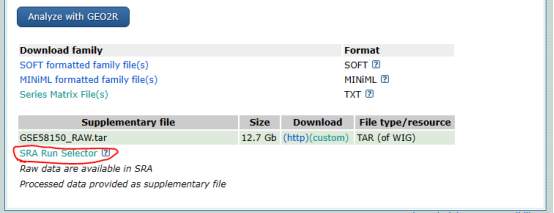
然后挑选你想要的样本或是选择所有样本,下载Accession List,
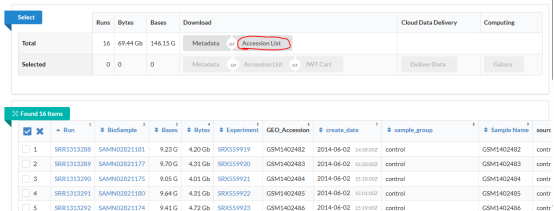
文件下载下来是这个样子的,里面是您选择的样本的SRR号
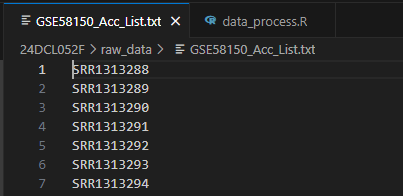
然后在服务器上运行命令:prefetch –option-file GSE58150_Acc_List.txt这样可以批量下载数据。
这样方便是方便,但是速度太慢了,一寸光阴一寸金,特别是对于科研工作者。这时候大家可以用ascp来下载数据,下面小花就教大家如何使用ascp来下载数据。
首先我们需要这个BioProject
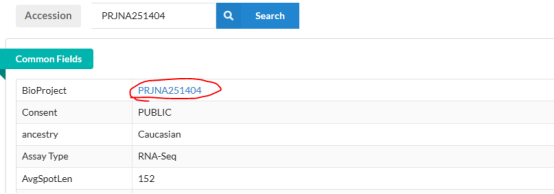
然后去ENA数据库搜索:


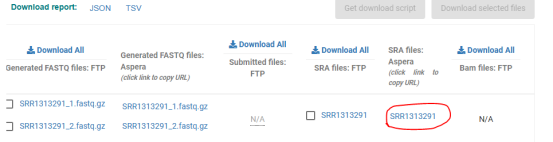
这样就复制下来我们的目标下载地址了:era-fasp@fasp.sra.ebi.ac.uk:/vol1/srr/SRR131/001/SRR1313291
然后使用命令:
ascp -P 33001 -k 1 -l 200m -T -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:/vol1/srr/SRR131/001/SRR1313291 ./
这里建议把所有下载地址都整理到一个文件中,比如这样:
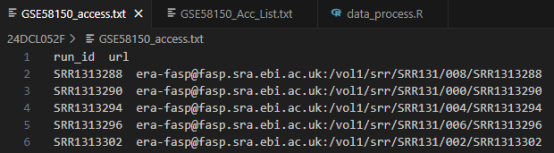
然后用for循环来逐个读取下载,但是有时候ascp会有下载失败的情况,这时候就需要重新下载。听上去是不是很麻烦你,别担心,小花已经写好了自动下载和自动重新下载的代码:
GSE_id = “GSE58150”
url = read.table(paste0(“./”, GSE_id, “_access.txt”), header = TRUE)
for(i in 1:nrow(url)){
system(paste0(“ascp -P 33001 -k 1 -l 200m -T -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh “,
url[i, 2], ” ./raw_data/”, GSE_id, “/”))
}
exist_file = list.files(paste0(“./raw_data/”, GSE_id, “/”))
fail_file = which(!url$run_id %in% exist_file)
while(length(fail_file!=0)){
for(i in fail_file){
system(paste0(“ascp -P 33001 -k 1 -l 200m -T -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh “,
url[i, 2], ” ./raw_data/”, GSE_id, “/”))
}
exist_file = list.files(paste0(“./raw_data/”, GSE_id, “/”))
fail_file = which(!url$run_id %in% exist_file)
}
把文件下载下来之后还要进行处理,别担心,这里小花也整理好了流程化的代码,您只需要创建几个文件夹然后设置几个路径就可以进行完所有流程:
samples = list.files(paste0(“./raw_data/”, GSE_id))
fastq_dir = paste0(“/media/desk16/iyunwf/ascp_down_test/”)
fastp_dir = paste0(“./fastp_data/”)
salmon_dir = paste0(“./salmon_result/”)
sra_dir = paste0(“./raw_data/”, GSE_id, “/”)
salmon_index = “./ref/GRCh38_salmon_index”
for(i in 1:20){
sra_file = paste0(sra_dir, “/”, samples[i])
system(paste0(“fastq-dump –gzip –split-3 –defline-qual ‘+’ –defline-seq ‘@\\$ac-\\$si/\\$ri’ “, sra_file, ” -O “, fastq_dir))
system(paste0(“rm “, sra_file))
system(paste0(“fastp -i “, fastq_dir, “/”, samples[i], “_1.fastq.gz “, “-I “, fastq_dir, “/”, samples[i], “_2.fastq.gz “,
“-o “, fastp_dir, “/”, samples[i], “.R1.fq.gz -O “, fastp_dir, “/”, samples[i], “.R2.fq.gz “,
“–thread 5”))
system(paste0(“rm “, fastq_dir, “/”, samples[i], “_1.fastq.gz “, fastq_dir, “/”, samples[i], “_2.fastq.gz”))
system(paste0(“salmon quant -i “, salmon_index, ” -l A “,
“-1 “, fastp_dir, “/”, samples[i], “.R1.fq.gz -2 “, fastp_dir, “/”, samples[i], “.R2.fq.gz “,
“-p 8 –validateMappings -o “, salmon_dir, “/”, GSE_id, “/”, samples[i], “_quant”))
system(paste0(“rm “, fastp_dir, “/”, samples[i], “.R1.fq.gz “, fastp_dir, “/”, samples[i], “.R2.fq.gz”))
}
把数据处理完之后我们还要把数据整合一下,这里也可以用小花的代码一次完成:
GSE_id = “GSE58150”
salmon_dir = paste0(“./salmon_result/”, GSE_id, “/”)
samples = list.files(salmon_dir)
library(stringr)
samples = str_extract(samples, “(.*?)_quant”, group = 1)
files = paste0(salmon_dir, “/”, samples, “_quant/quant.sf”)
for(i in 1:length(files)){
tmp = read.table(files[i], header = TRUE)
tmp = tmp[,c(1, 5)]
rownames(tmp) = tmp[,1]
colnames(tmp)[2] = samples[i]
if(i == 1){
merged_data = tmp
}else{
merged_data = cbind(merged_data, tmp[,2, drop = FALSE])
}
}
merged_data$ENST_id = str_extract(merged_data$Name, “(.*?)\\.”, group = 1)
write.csv(merged_data, paste0(“./data/data_”, GSE_id, “.csv”))
接下来就是正常的基因注释,重复基因取均值的步骤了。相信大家已经做的很熟练了。
做数据挖掘一定要熟悉各个数据库的使用,而且要熟悉数据库里各种数据类型,除了要有过硬的基础知识,一个好的工具也是必不可少的,工欲善其事必先利其器,要做这些操作,你要有一个服务器,空间够大,内存够大,线程够多,欢迎大家联系小花租赁高性价比的服务器哦。好了,今天的知识就分享到这里了,希望对大家能有所帮助,如果大家有其他疑问或者发现文中有不对的地方,欢迎留言分享。如果各位觉得自己运行代码太麻烦,欢迎用我们的云生信小工具(http://www.biocloudservice.com/home.html),只要输入合适的数据就可以直接出想要的图呢。