大家好!今天,我非常激动地向大家介绍生物信息学领域的一项强大工具——genefu 包。genefu包为基因表达分析提供了相关功能,尤其是在癌症中。该软件包包括许多用于分子亚型分类的算法。该包还包括预后预测算法的实现,以及这些算法所基于的预后基因签名列表。练了十年生信的我要凭本事吃饭了,要是您有自己做不了的生信分析,可以联系我。
在今天的学习中,让我们一起深入探索 genefu 包的使用方法。我们能够通过该包,进行分子亚型的分类、计算风险评分(Risk score)等。genefu 包不仅能够提升我们的数据分析能力,还能让我们可以更好地理解癌症中的数据,并将其生动地展示出来。希望大家能够积极学习和应用 genefu 包,不断探索新的工具和技术,提升自己在生物信息学领域的能力和水平。让我们一起开启学习之旅,共同探索生物信息学的奥秘吧!
本次介绍的R包需要较多的硬件资源,在服务器可以更加流畅运行,同学们如果没有自己的服务器欢迎联系我们使用服务器租赁~
分子分型
先给不熟悉癌症研究的小伙伴们介绍一下,癌症具有异质性,也就是说同一个部位患癌的癌症种类可能完全不一样,自然治疗方法也不一样。我们以下将使用的示例数据来自乳腺癌,在乳腺癌领域,不同亚型的癌症比不同器官来源癌症的差异要大很多。因此有必要对它进行分类,当然分类方法有很多,可以根据是否会转移分为恶性与良性。但在此我们使用分子分型,根据某些特征基因的表达量进行分类。
最简单的分子分型,当然是一个基因,比如ER阳性或者ER阴性的乳腺癌患者。专家共识建议采用标准流程检测乳腺原发灶和转移灶ER(Estrogen receptor,雌激素受体)、PR(Progesterone receptor ,孕激素受体)、HER2(Human epidermal growth factor receptor 2,人类表皮生长因子受体2)等,如果这3个基因都不表达,就是临床里面比较恶性的三阴性乳腺癌。另外还要一个增殖指数Ki67。并不是24有16种类型,而是五种,见《中国抗癌协会乳腺癌诊治指南与规范(2024年版)》:
管腔A型(Luminal A):ER/PR阳性且PR高表达HER2阴性,Ki-67增殖指数低;
管腔B型(Luminal B)HER2阴性:ER/PR阳性,HER2阴性且Ki-67增殖指数高或PR低表达;
管腔B型(Luminal B)HER2阳性:ER/PR阳性,HER2阳性(蛋白过表达或基因扩增),任何状态的Ki-67;
ERBB2(HER2又称ERBB2)阳性型:HER2阳性(蛋白过表达或基因扩增),ER阴性和PR阴性;
基底样型(Basal-like):三阴性(非特殊型浸润性导管癌),ER阴性,PR阴性,HER2阴性。
乳腺癌PAM50检测是目前临床应用中综合性价比最高的检测项目,它是(Parker et al., 2009)选定的有代表性的50个基因,利用它们可以对病人进行初步分类。根据基因表达谱(Gene expression profile, GEP)进行分型可称为分子分型。
genefu包介绍
genefu软件包包含实现基因表达分析通常需要的各种任务的功能,尤其是在癌症研究中:不同微阵列平台之间的基因图谱、分子亚型的鉴定、已发表基因签名的实现、基因选择和生存分析。这个包的使用简单直观,为生物信息学家和生态学家等领域的研究人员提供了强大的工具,帮助他们更好地理解和呈现系统发育树数据。
genefu包安装
需要R语言版本为4.4及以上,更低版本无法安装适合版本的BiocManager,会导致安装失败。在控制台中输入以下命令:
if (!require(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager”)
# 如果未安装BiocManager,则安装
BiocManager::install(“genefu”) # 安装genefu包
BiocManager::install(c(“breastCancerMAINZ”,”breastCancerTRANSBIG”, “breastCancerUPP”,”breastCancerUNT”,”breastCancerNKI”))
# 安装数据集
查看是否安装成功
packageVersion(“genefu”) # 查看genefu版本
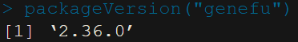
显示为2.36.0版本或更高,则表示已经安装了genefu包。
除此之外,后续示例还需要使用xtable,rmeta,Biobase,caret, survival等包,我们可以提前安装,安装命令如下:
install.packages(“xtable”)
install.packages(“rmeta”)
install.packages(“Biobase”)
install.packages(“caret”)
install.packages(“survival”)
使用genefu包进行分子分型
为了演示genefu包的用法,本文中我们将使用随机生成的树文件和模拟的关联数据集,使用的模拟数据集都包含树的那一列分类标签。
载入包与数据:
library(genefu)
library(xtable)
library(rmeta)
library(Biobase)
library(caret)
# 载入包
载入数据:
library(breastCancerMAINZ)
library(breastCancerTRANSBIG)
library(breastCancerUPP)
library(breastCancerUNT)
library(breastCancerNKI)
# 载入数据
数据概述如下表:
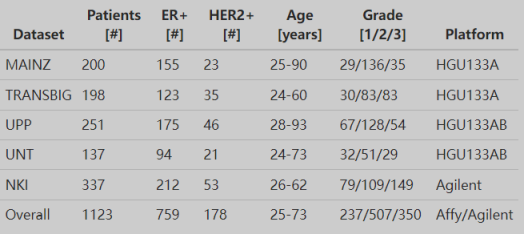
由表可知,五个数据集中各自的病人人数、ER阳性人数、HER2阳性人数、年龄、等级和数据采集所使用的平台。由于没有Ki-67增殖指数,所以只能按ER与HER2进行分类。ER阳性,HER2阴性为管腔A型(Luminal A);ER阳性,HER2阳性(蛋白过表达或基因扩增)为管腔B型(Luminal B);HER2阳性(蛋白过表达或基因扩增),ER阴性为ERBB2(HER2又称ERBB2)阳性型;ER阴性,HER2阴性基底样型(Basal-like)。
data(mainz)
View(mainz)
# 载入并查看mainz数据集
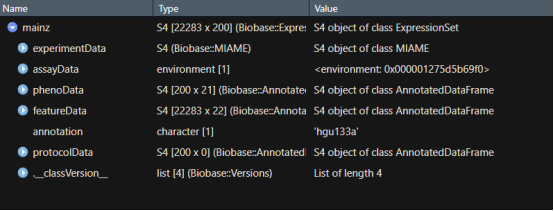
可见每个数据集都包括许多方面的数据,比如实验数据(experimentData)、表达数据(assayData)、样品信息(phenoData)、探针注释信息(featureData)、方法数据(protocolData)等。这其实是Biobase包中ExpressionSet类的格式,将多种来源信息组合成的单个数据结构,在芯片数据分析很常用。
data(breastCancerData)
# 载入示例数据集
cinfo <- colnames(pData(mainz7g))
# 使用mainz7g样品信息的列名
data.all<-c(“transbig7g”=transbig7g,”unt7g”=unt7g,”upp7g”=upp7g,”mainz7g”=mainz7g, “nki7g”=nki7g)
# 合并所有数据集
idtoremove.all <- NULL
duplres <- NULL
# 已知MainZ和NKI数据集没有重复,开始UNT、UPP与TRANSBIG的去重
demo.all <- rbind(pData(transbig7g), pData(unt7g), pData(upp7g))
dn2 <- c(“TRANSBIG”, “UNT”, “UPP”)
# 查找VDXKIU, KIU, UPPU系列
ds2 <- c(“VDXKIU”, “KIU”, “UPPU”)
demot<-demo.all[complete.cases(demo.all[,c(“series”)])&is.element(demo.all[ , “series”], ds2), ]
# 查找以上系列中重复的病人
duplid <- sort(unique(demot[duplicated(demot[ , “id”]), “id”]))
duplrest <- NULL
for(i in 1:length(duplid)) {
tt <- NULL
for(k in 1:length(dn2)) {
myx <- sort(row.names(demot)[complete.cases(demot[ , c(“id”, “dataset”)]) &demot[ , “id”] == duplid[i] & demot[ , “dataset”] == dn2[k]])
if(length(myx) > 0) { tt <- c(tt, myx) }
}
duplrest <- c(duplrest, list(tt))
}
names(duplrest) <- duplid
duplres <- c(duplres, duplrest)
# 查找VVDXOXFU, OXFU系列
ds2 <- c(“VDXOXFU”, “OXFU”)
demot <- demo.all[complete.cases(demo.all[ , c(“series”)]) &is.element(demo.all[ , “series”], ds2), ]
# 查找以上系列中重复的病人
duplid <- sort(unique(demot[duplicated(demot[ , “id”]), “id”]))
duplrest <- NULL
for(i in 1:length(duplid)) {
tt <- NULL
for(k in 1:length(dn2)) {
myx <- sort(row.names(demot)[complete.cases(demot[ , c(“id”, “dataset”)]) &demot[ , “id”] == duplid[i] &demot[ , “dataset”] == dn2[k]])
if(length(myx) > 0) { tt <- c(tt, myx) }
}
duplrest <- c(duplrest, list(tt))
}
names(duplrest) <- duplid
duplres <- c(duplres, duplrest)
# 全集中重复的病人
duPL <- sort(unlist(lapply(duplres, function(x) { return(x[-1]) } )))
# 加载数据
data(scmod2.robust)
data(pam50.robust)
data(scmgene.robust)
data(sig.ggi)
data(scmod1.robust)
data(sig.genius)
dn <- c(“transbig”, “unt”, “upp”, “mainz”, “nki”)
dn.platform <- c(“affy”, “affy”, “affy”, “affy”, “agilent”)
res <- ddemo.all <- ddemo.coln <- NULL
for(i in 1:length(dn)) {
# 加载数据
dd <- get(data(list=dn[i]))
# 删除重复项
message(“obtained dataset!”)
# 提取每个数据集的pData与fData
ddata <- t(exprs(dd))
ddemo <- phenoData(dd)@data
if(length(intersect(rownames(ddata),duPL))>0)
{
ddata<-ddata[-which(rownames(ddata) %in% duPL),]
ddemo<-ddemo[-which(rownames(ddemo) %in% duPL),]
}
dannot <- featureData(dd)@data
# 分子分型模型为PAM50,矩阵应样本为行,基因为列
PAM50Preds<-molecular.subtyping(sbt.model=”pam50″,data=ddata,annot=dannot,do.mapping=TRUE)
table(PAM50Preds$subtype)
ddemo$PAM50<-PAM50Preds$subtype LumA<-names(PAM50Preds$subtype)[which(PAM50Preds$subtype == “LumA”)] LumB<-names(PAM50Preds$subtype)[which(PAM50Preds$subtype == “LumB”)]
ddemo[LumA,]$PAM50<-“LumA”
ddemo[LumB,]$PAM50<-“LumB”
ddemo.all <- rbind(ddemo, ddemo.all)
}
table(ddemo.all$PAM50)
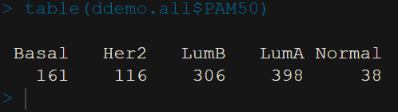
分子分型完成。
Normals<-rownames(ddemo.all[which(ddemo.all$PAM50 == “Normal”),])
ddemo.all$PAM50<-as.character(ddemo.all$PAM50)
# 比较了被PAM50模型预测为与分子分型有关的样本,忽视了‘Normal’的样本
confusionMatrix(factor(ddemo.all[-which(rownames(ddemo.all)%in%Normals),]$PAM50))
library(survival)
ddemo<-ddemo.all
data.for.survival.PAM50<-ddemo[,c(“e.os”,”t.os”,”PAM50″,”age”)] # 去掉缺失生存信息的病人
data.for.survival.PAM50<-data.for.survival.PAM50[complete.cases(data.for.survival.PAM50),]
days.per.month <- 30.4368
days.per.year <- 365.242
data.for.survival.PAM50$months_to_death<-data.for.survival.PAM50$t.os / days.per.month
data.for.survival.PAM50$vital_status<-data.for.survival.PAM50$e.os == “1”
surv.obj.PAM50<-survfit(Surv(data.for.survival.PAM50$months_to_death,data.for.survival.PAM50$vital_status)~data.for.survival.PAM50$PAM50)
message(“KAPLAN-MEIR CURVE – USING PAM50”)
# KAPLAN-MEIR 曲线(生存曲线) – PAM50模型
plot(main = “Surival Curves PAM50”, surv.obj.PAM50,col =c(“#006d2c”, “#8856a7″,”#a50f15”, “#08519c”, “#000000”),lty = 1,lwd = 3,xlab = “Time (months)”,ylab = “Probability of Survival”)
legend(“topright”,fill=c(“#006d2c”,”#8856a7″,”#a50f15″, “#08519c”,”#000000″),legend=c(“Basal”,”Her2″,”LumA”,”LumB”,”Normal”),bty = “n”)
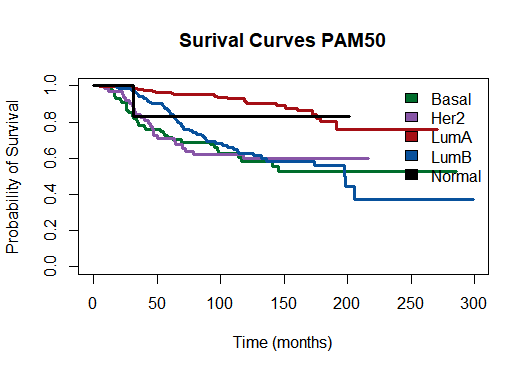
至此,我们不仅使用PAM50模型完成了分子分型,绘制了各型的生存曲线,更进一步挖掘了数据集的信息。
以上就是对于genefu包的全部介绍了,在本文中,我们详细介绍了如何使用 genefu 包来进行乳腺癌的分子分型,并对不同类型的乳腺癌预测了生存曲线。同学们可以通过学习和应用 genefu 包,掌握如何在 R 语言环境下进行癌症芯片数据分析,从而更好地理解和解释数据。当然这只是冰山一角,希望同学们继续深入学习和探索genefu 包的相关知识,不断尝试新的工具和技术,提升自己的数据分析能力和科研水平。
同学们如果觉得自己写代码麻烦,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表。立即访问云生信(http://www.biocloudservice.com/home.html),开启便捷的生信之旅!