嘿,大家好,欢迎伙伴们跟着小果一起探索生物信息学的奇妙世界。今天,小果将带领大家一起在DNA甲基化分析中探寻基因的奥秘,让我们一起看下去吧~
在过去的15年中,Infinium甲基化珠片微阵列已广泛用于全基因组DNA甲基化分析。继流行的EPICv1之后,新发布的EPICv2声称可以将基因组覆盖范围扩展到超过935,000个CpG位点。今天,我们要一起探索一个实战案例,在B细胞急性淋巴细胞白血病(ALL)和T细胞ALL样本间寻找EPICv2阵列中的差异甲基化区域(DMRs)。
在这个案例中,我们将使用DMRcate包,它是一个强大的工具,可以帮助我们在甲基化数据中找到那些差异甲基化区域(DMRs),就好像找到了基因的“指纹”,从而揭示出B细胞和T细胞急性淋巴细胞白血病样本间的独特差异,为我们的研究提供重要的生物学见解。小果提醒一句,这个R包操作占用内存比较大,建议使用服务器哦,欢迎联系小果租赁性价比高的服务器~
接下来,小果将带领大家一步步进行操作,从数据预处理,到DMRs的寻找,再到结果的解读,我们将一起揭开这个案例的神秘面纱。如果你在操作过程中遇到任何问题,别犹豫,记得联系小果哦~
我们先安装并加载DMRcate包:
if (!require(“BiocManager”))
install.packages(“BiocManager”)
BiocManager::install(“DMRcate”)
library(DMRcate)
第一步,获取数据。我们使用来自 Noguera-Castells 等人的样本子集来查找 T 细胞急性淋巴细胞白血病 (TALL) 和 B 细胞急性淋巴细胞白血病 (BALL) 之间的 DMR。我们从 ExperimentHub 加载 beta 矩阵:
library(ExperimentHub)
# 设置代理
#proxy <- httr::use_proxy(“http://my_user:my_password@myproxy:8080”)
## or
proxy <- httr::use_proxy(Sys.getenv(‘http_proxy’))
httr::set_config(proxy)
ExperimentHub::setExperimentHubOption(“PROXY”, proxy)
# 获取数据
eh <- ExperimentHub()
ALLbetas <- eh[[“EH9451”]]
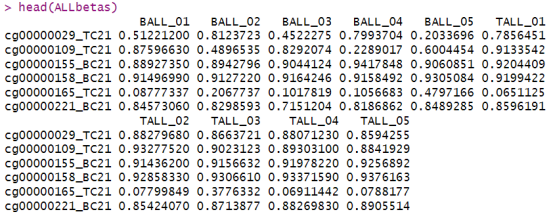
在输出中我们可以看到,ALLbetas数据中总共有10列数据,每一列代表一个样本的beta值,这10列分别代表 5 个 TALL 样本和 5 个 BALL 样本。
接下来,让我们绘制一个密度图,看一下ALLbetas数据集中的数据分布吧~ 跟着小果一起输入以下代码:
plot(density(ALLbetas[,1]), col=”forestgreen”, xlab=”Beta value”, ylim=c(0, 6),
main=”Noguera-Castells et al. 2023 ALL samples”, lwd=2)
invisible(sapply(2:5, function (x) lines(density(ALLbetas[,x]), col=”forestgreen”, lwd=2)))
invisible(sapply(6:10, function (x) lines(density(ALLbetas[,x]), col=”orange”, lwd=2)))
legend(“topleft”, c(“B cell ALL”, “T cell ALL”), text.col=c(“forestgreen”, “orange”))
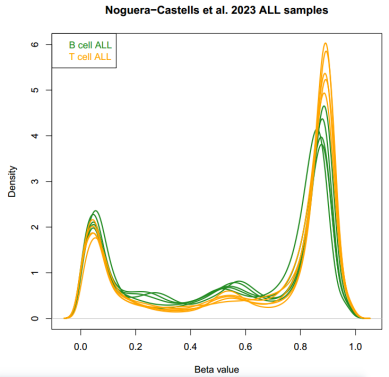
从密度分布图中可以看出,B细胞ALL和T细胞ALL的甲基化分布有所不同。这可能意味着这两种类型的白血病在甲基化水平上有所区别,这可能与它们的生物学特性和临床表现有关。然而,这只是一个初步的观察,我们需要进一步的统计分析来证实这一点。
现在让我们将 beta 值进行 logit2 变换,得到M值。这是因为M值近似于正态分布,更适合进行线性建模。
ALLMs <- minfi::logit2(ALLbetas)
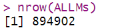
然后,打印出转换后的数据集ALLMs的行数,也就是得到探针的数量为894902。
nrow(ALLMs)
接着,使用rmSNPandCH函数过滤掉靠近SNP的探针。这里的rmcrosshyb = FALSE参数表示保留那些可能与基因组其他部分发生交叉反应的探针。
ALLMs.noSNPs <- rmSNPandCH(ALLMs, rmcrosshyb = FALSE)
最后,打印出过滤后的数据集ALLMs.noSNPs的行数,得到过滤后剩余的探针数量为893321。
nrow(ALLMs.noSNPs)
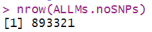
从输出结果可以看出,过滤后的探针数量比过滤前少了1581个,这些被过滤掉的探针可能是靠近SNP的探针。
与先前的版本EPICv1相比,EPICv2的新特性之一是它会有多个探针映射到同一CpG位点。这一特性在探针的性能测试上非常有用,但对于DMRcate则更希望探针到CpG位点是一对一映射的,否则核心将偏向那些具有更多复制品的位点。
rmPosReps()函数就可以调整M值矩阵以符合我们的要求。我们可以通过`filter.strategy`参数来指定选项。默认情况下,它会取每个复制组的平均值。
AnnotationHub::setAnnotationHubOption(“PROXY”, proxy)
ALLMs.noSNPs.repmean <- rmPosReps(ALLMs.noSNPs, filter.strategy=”mean”)
filter.strategy除了取平均值的方法,还提供了多种选择策略,如敏感度、精度或者随机选择。使用不同的参数,探针选择也会发生变化。下图显示了基于filter.strategy参数的探针选择的差异。
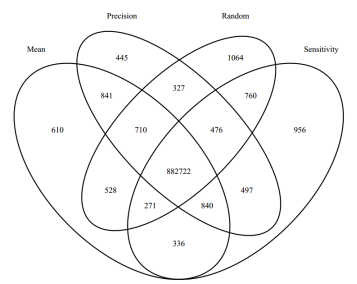
实际上,不同甲基化CpG位点的数量一般不太会因为filter.strategy参数的不同而有很大变化,但filter.strategy提供这种选择性,会有助于我们利用阵列上的所有可用探针。
接下来,让我们进入本次案例最重要的部分 —— 探寻差异甲基化区域(DMRs)!
首先,使用gsub函数从ALLMs.noSNPs.repmean的列名中提取样本类型。可以看到type是一个包含样本类型(”BALL”或”TALL”)的向量。
type <- gsub(“_.*”, “”, colnames(ALLMs.noSNPs.repmean))
接着,我们创建一个设计矩阵,用于后续的线性模型分析。PS:设计矩阵是一种常用于统计分析和机器学习的数据结构,它将解释变量(在这里是样本类型)编码为一组可以输入到模型中的数值。
design <- model.matrix(~type)

现在让我们来看看用于注释CpG位点的函数——cpg.annotate()。在这个函数中,逻辑参数epicv2Remap能允许我们在有证据显示探针更倾向于与脱靶CpG位点杂交的情况下,将探针重新分配到该位点。其中提到的证据一般来自于基因组比对和WGBS数据的一致性。当然,这仅适用于在rmSNPandCH()中指定了rmcrosshyb=FALSE的情况。
myannotation <- cpg.annotate(“array”, object=ALLMs.noSNPs.repmean, what = “M”,
arraytype = “EPICv2”, epicv2Remap = TRUE,
analysis.type=”differential”, design=design, coef=2,
fdr=0.001)

输出显示,在我们的案例中返回了29184个单独显著的探针。
接下来,我们可以使用dmrcate()函数来调用差异甲基化区域(DMRs)并对它们进行注释。需要注意的是,与之前的阵列不同,EPICv2阵列的注释是基于hg38版本的人类基因组的。
dmrcoutput <- dmrcate(myannotation, lambda=1000, C=2)
results.ranges <- extractRanges(dmrcoutput, genome = “hg38”)
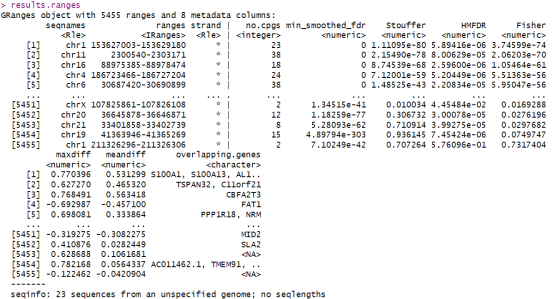
最后,我们可以使用DMR.plot()函数来绘制最重要的差异甲基化区域(DMRs)之一。
groups <- c(BALL=”forestgreen”, TALL=”orange”)
cols <- groups[as.character(type)]
DMR.plot(ranges=results.ranges, dmr=2, CpGs=ALLbetas,
what = “Beta”, arraytype = “EPICv2”, phen.col=cols, genome = “hg38”)
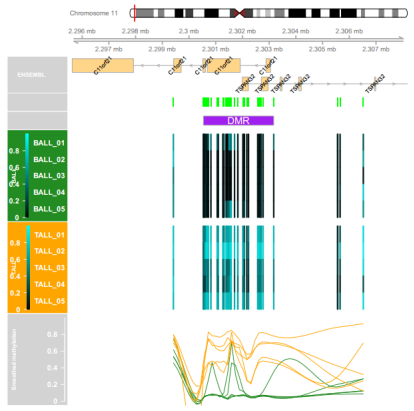
图中展示了特定DMR的甲基化水平,图由三部分组成:顶部显示DMR在第11号染色体的位置,中间部分展示每个个体在DMR的CpG位点的甲基化水平,底部显示每个组的平均甲基化水平。图分析表明,DMR可能与两组的表型差异有关,需要进一步研究。
伙伴们,小果已经带领大家一起完成了DMRcate的初步探索。现在,你可以开始自己动手,利用DMRcate包来分析差异甲基化区域(DMRs)啦~
无论你是在优化代码,还是在云端进行便捷的分析,云生信神器都能为你提供强大的支持。欢迎试试我们的云生信神器,只需一键上传数据,想要的图就能轻松get~