嘿朋友们,小果向导又来啦,欢迎来到生物信息学的奇妙世界。今天,我们要一起踏上一场阵列CGH(比较基因组杂交)数据的探索之旅。
阵列CGH,听起来是不是有点神秘?其实,这就是我们的超级侦探工具,能在基因组的海洋中寻找DNA的增加或缺失。这些微小的变化可能是疾病的罪魁祸首,比如让人闻风丧胆的癌症,或是神秘的神经发育障碍等等。阵列CGH就像一张宝图,上面的一系列对数比值就是线索,指引我们发现测试样本和参考样本在基因组各区域的DNA拷贝数的差异。这就像是一场寻宝游戏,你准备好加入我们,一起揭开基因组的神秘面纱了吗?
在这个旅程中,我们的得力助手是GLAD包。我们将用它来寻找那些隐藏在基因组深处的断点,并为每个神秘的染色体区域分配一个身份标签——正常、增加或缺失。注意哦,GLAD包操作占用内存比较大,建议使用服务器哦,欢迎联系小果租赁性价比高的服务器~
下面,小果将引领大家逐步掌握操作流程,从数据预处理开始,到寻找基因组断点,再到确定染色体区域的状态,我们将一同揭示阵列CGH数据分析的奥秘。如果你在旅途中遇到任何困难,别担心,小果随时待命,记得联系小果为你解答疑惑~
我们先安装并加载GLAD包:
if (!require(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager”)
BiocManager::install(“GLAD”)
require(GLAD)
在GLAD包中,阵列 CGH 谱分析有两个函数可用:glad 和 daglad。第二个函数是第一个函数的改进,因此更建议使用 daglad 函数。接下来,小果将为你演示这两个函数的使用方法。请注意,使用选项 smoothfunc=haarseg可以加快计算速度哦~
- glad函数
第一步,获取数据。我们使用来自 Snijders 等人 (2001) 的公共数据集。数据对应于 NIGMS 人类遗传学细胞库 (http://locus.umdnj.edu/nigms) 中 15 种已知核型的人类细胞株 (12 种成纤维细胞株、2 种绒毛细胞株、1 种淋巴母细胞株)。 每种细胞株均在 2276 个 BAC 的 CGH 阵列上进行杂交,并重复三次。加载数据后,需要将gm13330细胞系的数据创建为profileCGH对象。
data(snijders)
# 创建profileCGH对象
profileCGH <- as.profileCGH(gm13330)
接着,我们可以使用glad方法对细胞系gm13330进行分析,其中包括一系列参数的设置,如平滑函数、带宽、模型、核函数、基线调整等。
# 使用GLAD函数对数据进行处理
res <- glad(profileCGH, mediancenter=FALSE,
smoothfunc=”lawsglad”, bandwidth=10, round=1.5,
model=”Gaussian”, lkern=”Exponential”, qlambda=0.999,
base=FALSE,
lambdabreak=8, lambdacluster=8, lambdaclusterGen=40,
type=”tricubic”, param=c(d=6),
alpha=0.001, msize=2,
method=”centroid”, nmax=8,
verbose=FALSE)
最后,我们可以加载细胞带数据,并绘制分析结果图。
data(cytoband)
plotProfile(res, unit=3, Bkp=TRUE, labels=FALSE, plotband=FALSE, Smoothing=”Smoothing”, cytoband = cytoband)
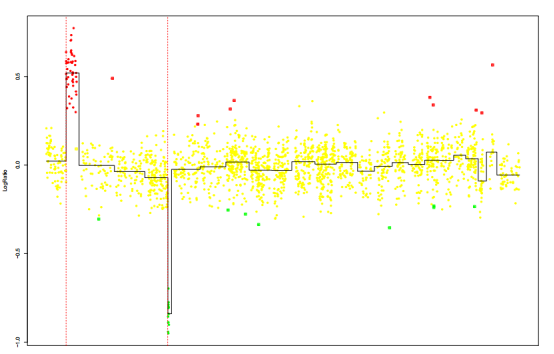
上图中展示了细胞系gm13330的DNA拷贝数变异分析结果,具体解释如下:
- 纵轴(log2比值):表示DNA拷贝数的对数比值。
- 横轴:表示基因组位置。
- 颜色点:
- 红色点:表示显著的拷贝数增加(增加)。
- 绿色点:表示显著的拷贝数减少(缺失)。
- 黄色点:表示没有显著变化的区域。(正常)
- 黑色线条:表示平滑后的拷贝数变异趋势。
- 红色虚线:表示断点位置。
通过这些信息,可以直观地看到基因组中哪些区域存在显著的拷贝数变异,这对于理解基因组结构变异和相关疾病研究具有重要意义。
- daglad函数
daglad函数的使用方法基本和glad一致哦,让我们快速过一遍吧!我们先加载来自Veltman 等人 (2003) 的数据,其中包括两个阵列 CGH 谱。
data(veltman)
profileCGH <- as.profileCGH(P9)
profileCGH <- daglad(profileCGH,mediancenter=FALSE,normalrefcenter=FALSE, genomestep=FALSE,smoothfunc=”lawsglad”,lkern=”Exponential”,model=”Gaussian”, qlambda=0.999, bandwidth=10, base=FALSE, round=1.5, lambdabreak=8, lambdaclusterGen=40, param=c(d=6), alpha=0.001, msize=2, method=”centroid”, nmin=1, nmax=8, amplicon=1, deletion=-5,deltaN=0.2,forceGL=c(-0.3,0.3),nbsigma=3,MinBkpWeight=0.35, CheckBkpPos=TRUE)
# 绘制结果图
plotProfile(profileCGH, Smoothing=”Smoothing”, Bkp=TRUE, plotband=FALSE, cytoband = cytoband)
abline(h=c(-0.3,-0.2,0.2,0.3),col=c(“green”,”black”,”black”,”red”))
axis(2,at=c(-0.3,-0.2,0.2,0.3), labels=c(“forceGL[1]”,”-deltaN”,”+deltaN”,”forceGL[2]”), las=2)
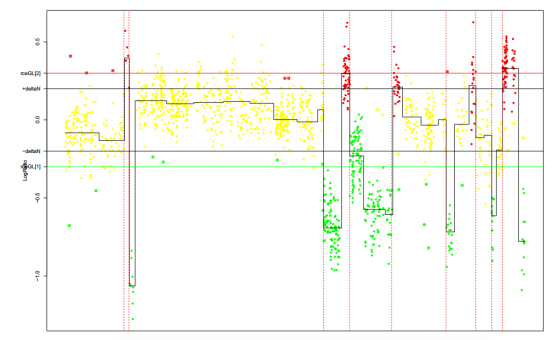
值得注意的是,daglad 函数允许选择一些阈值来帮助算法识别基因组区域的状态。阈值在以下参数中给出:deltaN、forceGL、deletion、amplicon。我们可以修改一下参数,比较一下不同参数对结果的影响。让我们把deltaN从0.2改到0.1,deltaN从c(-0.3,0.3)改到c(-0.15,0.15),看一下结果会有什么变化吧!
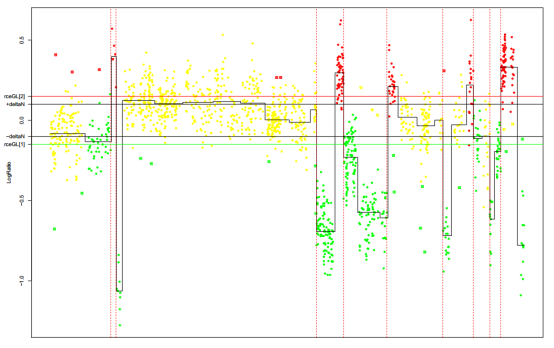
通过比较调整前后的结果图,可以更清楚地了解不同阈值对于算法结果的影响,以确定最适合数据集的参数设置。这种分析有助于优化算法的性能,并提高对基因组变异的识别能力。
在daglad函数中,如果设置了`genestep=TRUE`,则会对整个基因组进行平滑处理,连接所有染色体进行处理。算法会识别与正常DNA水平相对应的簇,然后将阈值与这些簇的中位数进行比较。这就可以根据整个基因组数据的全貌来调整阈值,以确保我们识别的变异区域在整个基因组中是显眼的,有助于提高我们算法的准确性和可靠性。
- 参数的调整
在上述函数中,比较重要的一些参数是`lambdabreak`、`lambdacluster`、`lambdaclusterGen`和`param`参数。通过调整这些参数,可以影响算法检测断点的数量。
- `lambdabreak`参数控制了算法中用于识别断点的惩罚力度,较小的值会导致更多的断点被检测出来。
- `lambdacluster`和`lambdaclusterGen`参数则用于控制簇(cluster)的识别,较小的值可能会导致更多的簇被识别出来。
- `param`参数中的`d`值也会影响算法检测断点的数量,较小的`d`值可能会导致更多的断点被检测出来。
针对信噪比非常小的阵列实验,建议降低上述参数的值,特别是将`param`参数中的`d`值调整为3或更小。这是因为在信噪比较小的情况下,对细微的变化更为敏感,因此降低参数值可以帮助算法在这种情况下更好地识别出潜在的断点,从而提高对变异的检测能力。
- 图形函数
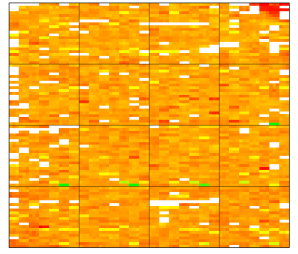
arrayPlot函数可以绘制出阵列 CGH 的空间图像,会显示不同基因组区域的信号强度比值,以便研究人员可以观察基因组的拷贝数变异等信息。
data(arrayCGH)
# object of class arrayCGH
array <- list(arrayValues=array2, arrayDesign=c(4,4,21,22))
class(array) <- “arrayCGH”
arrayPlot(array,”Log2Rat”, bar=”none”)
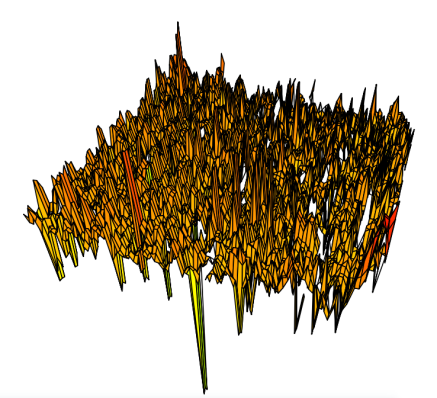
arrayPersp函数可以绘制阵列 CGH 三维透视图,展示了阵列比较杂交数据在空间中的分布情况。通过透视图,可以更直观地观察基因组的拷贝数变异、数据的分布规律以及不同区域之间的相对信号强度等信息。
arrayPersp(array,”Log2Rat”, box=FALSE, theta=110, phi=40, bar=FALSE)
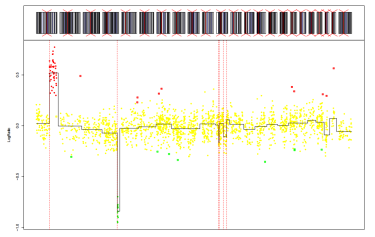
另外还有一个我们已经认识的老朋友——plotProfile函数,在上面介绍glad函数时,我们就已经使用过它来绘制整个基因组的图谱。如果想要在图中同时展示细胞遗传带,可以将plotband参数设置为TRUE(默认值)哦~
plotProfile(res, unit=3, Bkp=TRUE, labels=FALSE, Smoothing=”Smoothing”,plotband=TRUE, cytoband = cytoband)
除此之外,还可以用它来绘制1号染色体的基因组图谱和带标签的细胞遗传带,若不想同时绘制细胞遗传带,可以将plotband参数设为FALSE哦~
text <- list(x=c(90000,200000),y=c(0.15,0.3),labels=c(“NORMAL”,”GAIN”), cex=2)
plotProfile(res, unit=3, Bkp=TRUE, labels=TRUE, Chromosome=1,
Smoothing=”Smoothing”, text=text, cytoband = cytoband)
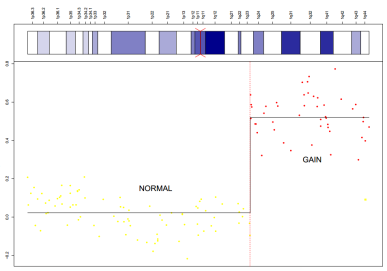
在这张图中,”NORMAL”表示染色体上正常区域,没有变异,生物特征正常。而”GAIN”表示基因组发生变异导致DNA拷贝数增加的区域。在这些区域,可能存在额外的基因拷贝或其他基因组突变,导致该区域的基因表达水平增加。这可能与肿瘤发展、细胞增殖等疾病相关,因此对”GAIN”区域的分析可以帮助研究相关疾病的发病机制和治疗策略。
伙伴们,小果已经带领大家一起完成了GLAD的初步探索。现在,你可以开始自己动手,利用GLAD包来分析阵列CGH数据啦,包括寻找基因组的断点、确定染色体区域的状态等。使用时记得充分利用GLAD提供的图形函数哦,可以让你的分析过程事半功倍!
无论你是在优化代码,还是在云端进行便捷的分析,云生信神器都能为你提供强大的支持。欢迎试试我们的云生信神器,只需一键上传数据,想要的图就能轻松get~