在做数据分析时,我们经常遇到需要for循环的程序,如果for循环内的操作简单还好,但是我们经常遇到在for循环内做复杂操作的程序,这种程序相当耗时,必须等前一个任务完成了才进行下一个任务,如果每个任务都与之前的任务无关,那我们就可以使用多线程来运行这个for循环。
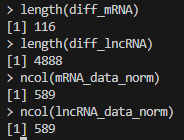
之前小花就遇到过计算mRNA与lncRNA相关性的分析,共有116个mRNA和4888个lncRNA,样本数是589,如果用普通的for循环的话那得运行到猴年马月,还好小花有服务器,可以在服务器上多线程运行。要是您有自己做不了的生信分析,可以联系我,如果您的数据量比较大也可以联系小花租赁高性价比的服务器哦。
首先我们来看一下数据:
原本小花也是用普通的for循环跑的,本来想着想让程序运行着,我去吃个饭再回来看,结果吃完饭又登陆几个小时程序还没有运行好:
up_glycolysis = read.table(“./input/up_glycolysis.txt”, sep = “\n”)$V1
down_glycolysis = read.table(“./input/down_glycolysis.txt”, sep = “\n”)$V1
diff_mRNA = c(up_glycolysis, down_glycolysis)
diffLab = read.csv(“./input/LUAD_lncRNA_diff.csv”)
#diffLab = diffLab[-which(is.na(diffLab$X)),]
diff_lncRNA = diffLab$X
mRNA_data_norm = read.csv(“./input/LUAD_mRNA_norm.csv”, row.names = 1)
lncRNA_data_norm = read.csv(“./input/LUAD_lncRNA_norm.csv”, row.names = 1)
m_lnc_cor = matrix(0, nrow = length(diff_mRNA), ncol = length(diff_lncRNA))
m_lnc_cor_p = matrix(0, nrow = length(diff_mRNA), ncol = length(diff_lncRNA))
rownames(m_lnc_cor) = diff_mRNA
colnames(m_lnc_cor) = diff_lncRNA
rownames(m_lnc_cor_p) = diff_mRNA
colnames(m_lnc_cor_p) = diff_lncRNA
for(i in 1:length(diff_mRNA)){
for(j in 1:length(diff_lncRNA)){
m_lnc_cor[i, j] = cor(as.numeric(mRNA_data_norm[diff_mRNA[i],]), as.numeric(lncRNA_data_norm[diff_lncRNA[j],]))
m_lnc_cor_p[i, j] = cor.test(as.numeric(mRNA_data_norm[diff_mRNA[i],]), as.numeric(lncRNA_data_norm[diff_lncRNA[j],]))[[3]]
}
}
这时候我想到,既然我的for循环内的语句前后间的计算结果并不会有影响,所以我想到了用多线程来运行:
## 多线程运行
library(parallel)
library(foreach)
library(doParallel)
# 自定义合并函数
combine_results <- function(…) {
# 将…中的所有列表转换为矩阵的列
lists <- list(…)
result <- do.call(rbind, lapply(lists, function(lst) unlist(lst)))
return(result)
}
# 检测可用的核心数
numCores <- detectCores()
# 创建并注册集群
clus <- makeCluster(numCores)
registerDoParallel(clus)
start_time <- Sys.time()
results <- foreach(i = 1:length(diff_mRNA), .combine=’combine_results’, .packages = c(“stats”)) %dopar% {
temp_cor <- numeric(length(diff_lncRNA))
temp_cor_p <- numeric(length(diff_lncRNA))
for(j in 1:length(diff_lncRNA)) {
temp_cor[j] <- cor(as.numeric(mRNA_data_norm[diff_mRNA[i],]), as.numeric(lncRNA_data_norm[diff_lncRNA[j],]))
temp_cor_p[j] <- cor.test(as.numeric(mRNA_data_norm[diff_mRNA[i],]), as.numeric(lncRNA_data_norm[diff_lncRNA[j],]))$p.value
}
list(cor = temp_cor, p_value = temp_cor_p)
}
end_time <- Sys.time()
# 关闭集群
stopCluster(clus)
time_taken_multi <- end_time – start_time
print(time_taken_multi)
m_lnc_cor = results[,1:length(diff_lncRNA)]
m_lnc_cor_p = results[,(length(diff_lncRNA)+1):(2*length(diff_lncRNA))]
rownames(m_lnc_cor) = diff_mRNA
colnames(m_lnc_cor) = diff_lncRNA
rownames(m_lnc_cor_p) = diff_mRNA
colnames(m_lnc_cor_p) = diff_lncRNA
saveRDS(m_lnc_cor, “./output/m_lnc_cor.rds”)
saveRDS(m_lnc_cor_p, “./output/m_lnc_cor_p.rds”)
这是用htop命令查看后台运行状况的结果,可以看到56个线程全部占满了:

最后只用了11分钟就结束了战斗:

这大大提高了我们的分析效率,当然不是所有程序都能使用多线程来运行,只有具备这些特点的程序才能改写成多线程运行的方式:
1、任务可分割性:程序中的任务可以被分割成多个小任务,这些小任务可以独立执行而不会相互干扰。
2、数据依赖性低:理想的多线程任务应该具有较低的数据依赖性,这意味着不同线程处理的数据可以相对独立,减少了线程间的通信和同步需求。
聪明的你也许已经发现了,我们的程序中使用了foreach和%dopar%,因为在 R 语言中,foreach 循环通常与 %dopar% 结合使用,以实现并行计算。这样可以显著提高处理大数据集时的效率,因为它允许同时在多个核心上执行代码。这是在 R 中使用 foreach 而不是普通 for 循环的主要原因之一。%dopar% 是 R 语言中 foreach 包的一个操作符,用于在并行计算中执行循环。当你使用 %dopar% 时,它会将 foreach 循环中的每次迭代分配到不同的核心或处理器上并行执行,这样可以显著提高计算效率,特别是在处理大量数据时。
代码中的combine_results是我们自定义的合并函数,用于将多线程计算的结果合并,因为我们同时计算了相关性和相关性pvalue,所以不能用普通的rbind或cbind合并。
好了,今天的知识就分享到这里了,如果各位觉得自己运行代码太麻烦,欢迎用我们的云生信小工具(http://www.biocloudservice.com/home.html),只要输入合适的数据就可以直接出想要的图呢。一寸光阴一寸金,寸金难买寸光阴,服务器可以大大提升我们的工作效率,欢迎联系小花租赁高性价比的服务器哦。