
嗨,我是小师妹,很高兴能够和大家一起来学习生物信息学。如果说老师让你去找两个个体的基因组变异情况,相信很多人都知道先选参考基因组进行BWA比对,得到bam文件后使用GATK或samtools软件等等一套流程得到VCF文件,最后进行注释就得到基因组之间的变异情况了。但这是有原始数据的情况,那假设我们是想比较两条基因组文件的差异呢?手里没有raw data又如何进行基因组变异检测呢?本次介绍的软件操作占用内存比较大,建议使用服务器,欢迎联系小师妹租赁性价比居高的服务器~
基因组共线性神器MUMmer4这就来啦,MUMmer能够用于研究细菌不同菌株间的基因组倒位现象,分析大鼠与小鼠基因组在进化过程中的重排,以及比较同一物种不同基因组组装版本的差异等。其基于后缀树算法,其运行速度远超BLASTZ(基于k-mers)。后缀树(Suffix tree)是一种数据结构,能快速解决很多关于字符串的问题。
MUMmer是一种快速比对大DNA序列的系统。它可以对齐:
- 全基因组到其他基因组
- 大基因组相互组装
- 部分(草稿)基因组序列相互连接
- 或者(在版本4中)对基因组进行一组读取。
前几天小师妹给大家带来了系统发育分析一站式的分享!今天给大家介绍的是一个非常强大基因组共线性神器工具——MUMmer,同时也可以做SNP检测。遗传分析可难不倒生信高手小师妹,要是同学们有自己做不了的生信分析,欢迎随时联系我!!!

一、下载和安装
mummer发表于1999年,是经典的全局比对软件。一直有更新维护,最新的版本是2020年10月发布的4.0.0 candidate 1版本,下面就给大家带来最新鲜的mummer4安装指南:

图1 Mummer工具各版本更新情况
首先我们需要先安装所需的REQUIREMENTS,建议新建一个conda环境
- perl5 (5.6.0)
- sh
- sed
- awk
OPTIONAL UTILITIES
- fig2dev (3.2.3)
- gnuplot (4.0)
- xfig (3.2)
接下来是安装部分:
$ conda create -n mummer4
$ conda activate mummer4
$ conda install -c conda-forge perl
$ conda install -c conda-forge fig2dev
$ conda install -c conda-forge xfig
$ mkdir mummer4
$ cd mummer4
$ wget https://github.com/mummer4/mummer/releases/download/v4.0.0rc1/mummer-4.0.0rc1.tar.gz
$ tar -zxvf mummer-4.0.0rc1.tar.gz
$ ./configure –prefix=/path/to/installation
$ make
$ make install
$ export PATH=$current_path/mummer-4.0.0rc1:$PATH
Mummer一共有4个工作流程:
- nucmer: 由Perl写的流程,用于联配很相近(closely related)核酸序列。它比较适合定位和展示高度保守的DNA序列。注意,为了提高nucmer的精确性,最好把输入序列先做遮盖(mask)避免不感兴趣的序列的联配,或者修改单一性限制降低重复导致的联配数。
- promer:也是Perl写的流程,它以翻译后的氨基酸序列进行联配,工作原理同nucmer。
- run-mummer1,run-mummer3:两者是基于cshell写的流程,用于两个序列的常规联配,和promer,nucmer类似,只不过能够自动识别序列类型。它们擅长联配相似度高的DNA序列,找到它们的不同,也就是适合找SNP或者纠错。前者用于1v1无重排,后者1v多有重排
二、常用命令
1、比对
nucmer通过比较基因组获取突变信息,该方法适合多个近缘物种的基因组比较,可以用于多Fasta数据文件中包含的核苷酸序列的全对全比较。它最适合用于可能具有大重排的高度相似序列。常见的例子是:比较两个未完成的鸟枪式测序组件,将未完成的测序组件映射到完成的基因组,以及比较两个可能有大重排和重复的相当相似的基因组。
$ nucmer -p align ref.fasta contig.fasta
# nucmer -p [out_index] [ref] [query]
# -p|prefix:Set the prefix of the output files (default “out”)
# –mum: Use anchor matches that are unique in both the reference and query
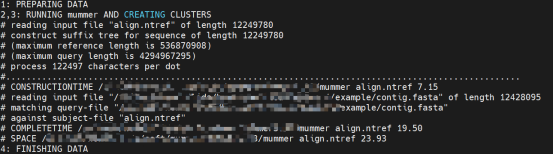
图2 比对过程
结果文件会生成一个delta为后缀的文件,让我们打开它看看:
$ less align.delta
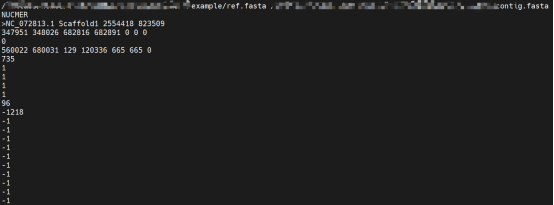
图3 nucmer比对结果文件
- 文件第一行是两个需要比较的基因组序列fasta文件的绝对路径。
- 第二行是比对类型,可以是NUCMER或PROMER。
- 第三行往后是比对结果。每个比对结果以行首为 > 号开头,整行为 0 结尾。
- 比对结果第一行以 > 号开头,后面包含空格分割的4列,分别是:reference序列 ID、query序列ID、该reference序列长度、该query序列长度。
- 比对结果第二行包含空格分割的7列数值,分别是:reference序列比对起始位点、 reference序列比对结束位点、query序列比对起始位点、query序列比对结束位点、错配位点数、不相似位点数、非字母字符个数。
- 第三行往后是Delta值,表示离下一个IDNEL之间的距离。
2、过滤
序列中的重复序列可能会掩盖可能的SNP,所以我们可以使用delta-filter去除一对多、多对多中的冗余匹配。这里的结果会比较晦涩难懂,我们后面可以进行可视化。
$ delta-filter -1 -i 90 -q -r align.delta > align.filter.delta
常用示例:
$ delta-filter -i 90 -l 2000 -m out.delta > out.f.delta
常用参数:
-i <float> default:0
该参数的值必须设定为0~100,表示过滤掉Identity低于此值的比对结果。
-l <float> default: 0
过滤掉匹配区域长度低于此值的比对结果。
-u <float> default: 0
要求比对区域的unique序列所占的百分比不低于此值。
-q
对query序列的每个比对位点,仅选择其在reference序列上的最优比对结果。
若需要将contigs序列或reads定位到reference genome上,考虑使用-q参数。
-r
对reference序列的每个比对位点,仅选择其在query序列的最优比对结果。
-g
加入该参数,则要求比对区域内部的maximal matches之间不能有重排,如
移位、倒位等。
-m
加入该参数,表示取-q和-r参数的并集。一个query位点可能比对到ref上多
个位点,仅保留其最优比对结果;一个ref位点也可能比对到query上多个位点,仅
保留其最优比对结果;以上两个处理方法的输入信息是一样的,都是所有的比对结果;
若使用 -q -r 参数,则是先运用第一种方法处理,得到的结果再运用第二种方法
处理,其结果相比于仅使用 -m 参数,输出的比对结果更少。一般情况下,使用 -m
参数是最优的方法。
-1
加入该参数,表示取-q和-r参数的交集。一个query位点可能比对到ref上多
个位点,一个ref位点也可能比对到query上多个位点;加入该参数,表示要求query
位点和ref位点能相互比对上,且它们是相互最佳比对;若使用 -q -r 参数,得到的
虽然也是1对1的比对结果,但其不一定是相互最佳比对,-1参数的输出对结果更少。
若需要得到SNP结果,则需要加入该参数来确保结果的准确性。

图4 过滤结果文件
3、坐标转换
使用show-coords脚本可以将delta文件转换为易读的匹配坐标,这一部分可以进行两个基因组的共线性分析。
$ show-coords -c -r align.filter.delta > align.filter.coords
#-c 输出结果中多处覆盖比这一列
#-r 对结果进行排序输出
结果文件为align.filter.coords,让我们来看一看结果如何:
$ less align.filter.coords
[S1]和[E1],第一个基因组中,和第二个基因组对齐的区域,S1和E1分别为这段区域在第一个基因组中的起始和结束位点;
[S2]和[E2],第二个基因组中,和第一个基因组对齐的区域,S2和E2分别为这段区域在第二个基因组中的起始和结束位点(如果起始位点比结束位点的数值大,则意味着在这段区域中和其互补链对齐);
[LEN1]和[LEN 2],分别两个基因组中,对齐区域的长度;
[% IDY],两个基因组中对齐区域的碱基一致性,完全一致则是100%;
[COV R]和[COV Q],覆盖度。
[TAGS],基因组中两段存在共线性区域的序列的名称;

图5 坐标转换结果文件
4、Call Snp
接下来我们可以使用show-snps显示过滤后的delta文件中匹配的SNP:
$ show-snps -C -I -T -r -l align.filter.delta > align.filter.snp
USAGE: show-snps [options] <deltafile>
-C 不要报告具有模糊映射的比对中的SNP,即只报告[R]和[Q]列等于0的SNP,而不输出这些列
-h 显示帮助信息
-H 不打印输出标题
-I 不报告indels
-l 在输出中包含序列长度信息
-q 按查询ID和SNP位置对输出行进行排序
-r 按参考ID和SNP位置对输出行进行排序
-S 通过向stdin传递“显示坐标”行来指定要报告的对齐方式
-T 切换到制表符分隔格式
-x int在输出中包含周围SNP上下文的x个字符,默认为0

图6 Show-snps结果文件
以上结果文件就是SNP结果文件啦,我们可以将其转化为VCF文件,再转换为矩阵,提取出fasta之后还可以进行全基因组SNP的系统发育分析~小师妹在往期的公众号也有提到系统发育分析的一站式流程,欢迎查阅。
三、小结
Mummer工具主要有两大应用场景,一是进行基因组之间的共线性检测,二就是全基因组范围的SNP检测,与常规gatk或bcftools的call snp流程不同,Mummer工具采用的是后缀树算法,速度方面会快很多。
系统发育相关的基因组之间既存在保守性又存在可变性。有些序列片段的数目以及顺序具有保守性,这种保守性可以使用共线性来进行描述。共线性主要强调两方面,一是序列的同源性,二是序列片段的排列顺序。同时即使很近缘的基因组也可能存在大量的变异和多态性,这种变异可能构成了不同个体与群体性状差异的基础。单核苷酸多态性(single-nucleotide polymorphism,SNP)是指由于单个核苷酸位置上存在转换或颠换等变异所引起的DNA序列多态性,常用来研究近缘物种基因组的进化。
今天小师妹给大家重点介绍的是SNP检测,当然大家也可以根据自己的需求和研究目标修改和扩展这些代码哦!!怎么样,你学会了嘛!?
同学们如果觉得自己写代码麻烦,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表。立即访问云生信(http://www.biocloudservice.com/home.html),开启便捷的生信之旅!