
嗨,我是小师妹,很高兴能够和大家一起来学习生物信息学。前面几天在外网上看到一个针对细菌基因组SNP鉴定工具的调查:

图1 SNP检测工具流
从图中可以看出snippy还是比较受欢迎的,接口也很简单。这次解读的主角是snippy,使用起来确实很方便,一条命令可以解决我们从sra到vcf到的问题,当然其中使用的工具很多了,但是都被封装在脚本里了。本次介绍的软件操作占用内存比较大,建议使用服务器,欢迎联系小师妹租赁性价比居高的服务器~
- 测序数据与参考基因组比对获得sam、bam数据
- Samtools、GATK、freebayes等软件利用bam文件获得VCF
- Snpeff对VCF文件进行注释
- Vcf文件转换成fasta格式使用IQtree、mega等软件构建系统发育树
Snippy 是一个用于快速发现单倍体参考基因组与下一代测序(NGS)序列读取之间单核苷酸多态性(SNPs)和插入/删除(indels)的工具。它设计用于在单个计算机上利用尽可能多的CPU核心,以实现高速处理。Snippy 由 Torsten Seemann 开发,是一个广泛应用于基因组变异分析的开源工具。下面小师妹就庖丁解牛般解读下这些流程工具是如何工作的,流程又是如何设计的,一起看下去吧。遗传分析可难不倒生信高手小师妹,要是同学们有自己做不了的生信分析,欢迎随时联系我!!!

一、下载和安装
1、安装部分
Conda安装
snippy=4.6.0为作者推荐的版本,而snippy=4.6.0兼容snpeff=5.0,更高的版本则可能报错。安装后,如果运行snippy时出现一些执行上的问题,需检查所依赖的软件版本。
$ conda install -c conda-forge -c bioconda -c defaults snippy
Mac安装
$ brew install brewsci/bio/snippy
Github安装最新版
$ cd $HOME
$ git clone https://github.com/tseemann/snippy.git
$ HOME/snippy/bin/snippy –help
# 添加snippy以及依赖库的路径到环境变量中,可直接调用snippy
$ echo ‘export PATH=”$HOME/repos/snippy/bin:$HOME/repos/snippy/binary/linux:$PATH”‘ >> ~/.bashrc
$ source ~/.bashrc
2、核验安装是否完成
检查安装软件的版本
$ snippy –version
检查所有依赖项是否已安装并正常工作
$ snippy –check
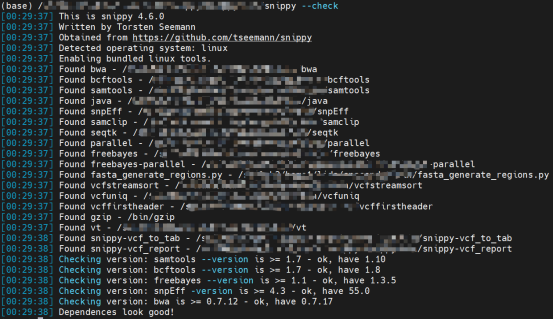
图2 检查依赖项
二、常用命令
1、Snippy-multi
Snippy共有三种输入方式,分别是fastq、拼接好的fasta以及比对好的bam文件
USAGE
snippy [options] –outdir <dir> –ref <ref> –R1 <R1.fq.gz> –R2 <R2.fq.gz>
snippy [options] –outdir <dir> –ref <ref> –ctgs <contigs.fa>
snippy [options] –outdir <dir> –ref <ref> –bam <reads.bam>
以上是单个文件的输入方式,如果你有多个文件,可以用snippy-multi命令:
# input.tab = ID R1 [R2]
Isolate1 /path/to/R1.fq.gz /path/to/R2.fq.gz
Isolate1b /path/to/R1.fastq.gz /path/to/R2.fastq.gz
Isolate1c /path/to/R1.fa /path/to/R2.fa
# 单端读取也支持
Isolate2 /path/to/SE.fq.gz
Isolate2b /path/to/iontorrent.fastq
# 或已经组装的元件,如果你没有读取数据
Isolate3 /path/to/contigs.fa
Isolate3b /path/to/reference.fna.gz
为了便于将多个测序数据比对到参考基因组,推荐使用 snippy-multi。这个脚本的输入文件需要由制表符(tab)分割,输入文件可以是单端、双端和组装的测序数据(paired-end reads, single-end reads, 和 assembled contigs)。按照github的说明,菌株和fasta之间有个Tab键就可以了。以下拿组装后的contig作为例子,这里的输入是七个组装好的contig文件:
$ less input.tab

图3 input.tab文件内容
然后运行以下命令来生成输出脚本。第一个参数应为 input.tab 文件。其余参数应该是共享的 snippy 参数。
$ snippy-multi input.tab –ref ref.fna –cpus 16 > runme.sh
检查脚本是否合理,最后一行它还会在最后运行 snippy-core 以生成核心基因组 SNP 对齐文件 core.*:
$ less runme.sh
图4 运行脚本
运行脚本:
$ sh ./runme.sh
结果文件成功生成了,让我们打开一个来看看:

图5 输出结果文件
相关的结果文件描述如下:

图6 相关文件描述
2、Snippy-core:core SNP系统发育分析
如果要将多个菌株比对到同一个参考基因组上,可以产生一个“core SNP”的比对,可用来建立具有高度分辨率的系统发育树(忽视可能的基因重组)。一个核心位点是指存在于所有细菌样本中的基因组位置。一个核心位点可以在每个细菌样本中具有相同的核苷酸,也可以不同。如果我们忽略细菌中的的插入和缺失突变的复杂性,仅使用变异位点,这些就是core SNP genome。其中Snippy-core的命令已经包含在了Snippy-multi的脚本中,
$ snippy-core –ref ref.fa case1 case2 case3 case4 case5 case6 case7
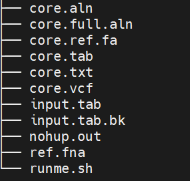
图7 结果文件
core.aln:这是一个核心基因组的比对文件,包含所有样本的核心基因组序列。用于后续的系统发育树构建和进化关系分析。
core.full.aln:包含所有样本的所有变异位点的比对文件,不仅仅是核心基因组部分。详细展示了每个样本中变异位点的信息。
core.tab:一个表格文件,详细列出了所有变异位点及其在各个样本中的状态。该表格可以用于进一步的统计分析和变异注释。
core.vcf:变异位点的 VCF(Variant Call Format)格式文件,广泛用于存储基因组变异信息。
该文件可以与其他生物信息学工具兼容,用于进一步的变异分析。
接下来我们可以利用core.aln进行全基因组SNP系统发育分析,利用我们上期分享过的IQTree2流程进行进化树构建。如果想要找最佳模型,可以用-m MF参数,这里用的是GTR模型,结果大差不差。
$ perl mfa2phy.pl core.aln core.aln.phy
$ nohup iqtree2 -s core.aln.phy -m GTR -bb 1000 -T AUTO &
然后后再将treefile文件放到进化树美化软件就可以看到结果啦,这里导入的是在线网站iTOL,我们也可以结合材料背景信息进行美化。
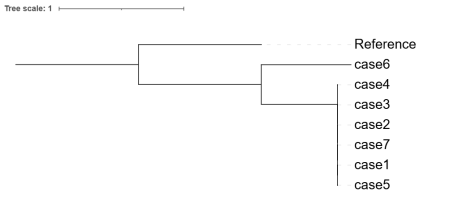
图8 全基因组SNP进化树
3、Snippy-dist:core SNP系统发育分析
当我们分析院内感染,菌株传播关系的时候。传统最常用的方法有16SRNA,MLST,cgMLST,以及全基因组的分析,分辨率依次升高。今天snp差异数就是基于全基因组分析得到的最高精度的菌株传播关系之间的证据。首先对自己的菌株的多个基因组进行比对生成.aln结尾的文件,然后就可以计算snp差异了。不同的菌株阈值不同,对于一般细菌菌而言SNP<40个被认为直接的菌株播散。
$ snp-dists core.aln > core.aln.snpmatrix

图9 中间处理过程
$ less core.aln.snpmatrix

图9 SNP矩阵文件
可以看到结果文件是一个SNP矩阵,接下来我们就可以用热图来展示啦。

图10 SNP矩阵热图可视化
从这个矩阵中,我们可以判断case3、case2和case4属于直接传播。
三、小结

图11 Snippy的核心功能
Snippy的核心功能见上图,但之所以说是细菌基因组遗传分析神器,原因在于该工具可以自动判别变异,熟悉人类的全基因组/全外显子组”家系”与”肿瘤”变异检测原理的读者们,应该能理解这个参数。此参数背后的概念是为单倍体生物(即:细菌、病毒、真菌、原生动物等低等生物)的遗传分析”量身定做”,否则将带来统计学和遗传学上的麻烦(GATK的基因型、Mutect2-GATK的突变负荷等统计概念与方法,在此处均不适用),以二倍体生物为例,还要需要进行杂合推断,而对于单倍体生物来说,不应该存在杂合子,也无“突变负荷”。
今天小师妹给大家重点介绍的是SNP检测,当然大家也可以根据自己的需求和研究目标修改和扩展这些代码哦!!怎么样,你学会了嘛!?
同学们如果觉得自己写代码麻烦,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表,上面的进化树、热图也可以轻松绘制。立即访问云生信(http://www.biocloudservice.com/home.html),开启便捷的生信之旅!