云生信学生物信息学
点击蓝字关注我们
癌组织与微生物的研究是当前癌症研究领域的新热点,大规模临床活检样本测序数据平台(如TCGA)的出现为癌症组织与微生物关联研究提供了相当大的便利,由于不同来源的数据往往存在批次效应,而且不可避免的在采样过程中产生污染,而如何在来自不同测序平台的数据中区分来自组织内定植的菌还是来源于污染的菌是当前癌组织与微生物关联研究面临的一大挑战。尽管目前有很多策略被应用到TCGA数据的去污染,但是结果如何从未验证过。在一篇2021年2月10日发表在Cell Host & Microbe的文章里,作者对TCGA中来自1772例患者的3,689个独特样本的4,937次测序数据(囊括了5个TCGA项目和21个解剖部位),运用算法分离并验证了其中污染来源的序列,将净化后的数据整理并释放到公共数据库(TCMA),并提供了下载(https://tcma.pratt.duke.edu)。同时,作者运用净化后的数据确定了结肠直肠癌(CRC)组织中比血液里更倾向于定植与结肠粘膜屏障损伤相关的菌,我们一起来看一下作者是如何一步步识别污染序列并将其净化的。

1、全基因组测序(WGS)和全外显子测序(WXS)显示CRC携带的大肠细菌与血液和大脑不同
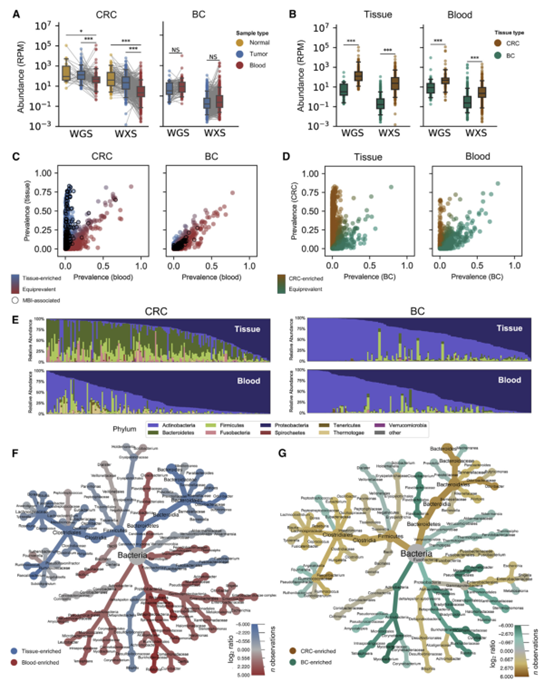
作者最先分析了来自不同测序平台的617名结肠癌病人的730份癌组织和555份血液样本以及923名脑癌病人的958份癌组织和914份血液样本的WGS和WXS数据,发现CRC组织和血样微生物丰度的比较分析揭示了两种不同的细菌组成:一种是富集于组织的菌,另一种普遍存在于组织和血液中的菌。在实体组织中微生物丰度比来自对应个体的CRC患者血液样本中更丰富、更多样。相比之下,在大脑癌症(BC)病人中,微生物的丰度和多样性在组织中比匹配的血液样本中差别并不大。同时CRC组织的微生物群落比BC组织更丰富,更多样。而且在CRC组织相比于CRC病人血液样本有更多的已报道的肠粘膜损伤相关菌,相反的,来自BC病人的组织和血液样本的菌几乎相当,只有少数富集在BC组织。拟杆菌门(Bacteroidetes),厚壁菌门(Firmicutes)和梭杆菌门(Fusobacteria)在CRC组织中的比例比较高,变形菌门(Proteobacteria)和放线菌门(Actinobacteria)在CRC血液样本和BC组织样本中所占比例相对较高(如图)。
2、在组织和血液中普遍存在的物种主要来源于污染物
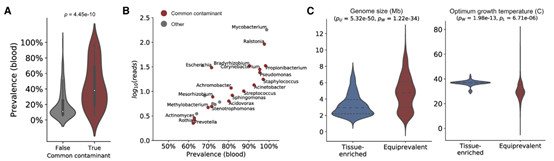
除了富集在CRC组织的菌群外,在血液、CRC组织、BC组织和OV组织中都检测到一些普遍存在的物种。作者认为受损的上皮屏障功能可能使微生物以低水平易位进入血流,作者找了70种普遍存在于健康人宏基因组数据种的菌,然后发现这些菌在血液中更富集。在CRC组织和血液中普遍存在的物种在G-C含量、基因组大小和最佳生长条件方面也比在CRC中富集的物种具有更大的遗传和表型多样性。相反,CRC富集的物种对极端生长条件的耐受性要差得多,其最佳温度、pH值和NaCl水平更接近于人类的内稳态。作者认为,那些普遍存在组织和血液中的菌因为有更大的基因组,比肠道微生物群更能忍受多变和不稳定的环境条件而更可能是来自污染。
3、在血液和组织中普遍的物种与特定的测序中心有关
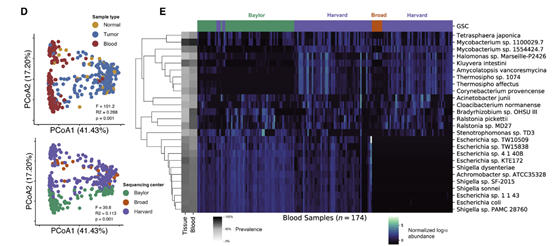
CRC样本WGS数据的PCoA显示,血液样本和组织样本沿第一个变异轴存在相当大的差异主要反应样本组织的差异,而批效应沿第二个轴存在,有趣的是反映了测序中心的差异(如下PCOA图),血液样本细菌种类丰度的差异也显示测序中心特异性污染的存在(如下图)
4、作者开发了一种用于分离TCGA肿瘤样本中组织常驻菌群的可推广模型
基于组织和血液中流行率的比较分析,作者开发了一个可推广的统计模型来区分TCGA中不同癌症类型的组织内微生物群和污染物物种。在5%以上的CRC组织样本中检测到1136种细菌,该模型将769种细菌划分为组织滞留菌(67.69%),367种细菌划分为污染物(32.31%)。通过比较组织和血液中的流行率确定的组织居民人群与CRC组织和BC组织的流行率比较以及与WXS数据进行的类似比较基本一致
5、来自变形菌门和放线菌门的菌在污染物读量中所占比例最大
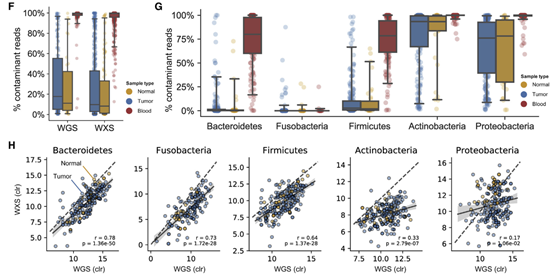
作者应用算法净化序列后统计在WGS数据中,组织中污染来源的序列占比中位数为16.27%,血液中污染来源序列占比中位数则为99.45%。CRC组织内,变形菌门和放线菌门对污染序列的贡献率最大,其中位数分别为76.67%和80.95%。在匹配的WGS和WXS样本中,拟杆菌门(Bacteroidetes)、梭杆菌门(Fusobacteria)和厚壁菌门(Firmicutes)的丰度相关性一直较高,而放线菌门(Actinobacteria)和变形菌门(Proteobacteria)的丰度相关性较低。总的来说,这些结果表明,CRC组织和血液样本中显著的比例是来自放线菌和变形菌种污染。
6、基因水平检测组织常驻和污染物物种
正如预期的那样,组织和血液样本的基因分布在很大程度上重现了种水平的分布。对于组织定植的物种,其在血液样本的基因比例远低于组织样本,而对于污染物种,血液样本和组织样本的基因比例几乎相同。对于组织定植的物种,其组织中的基因组覆盖度要大于其在血液中的覆盖度,但对于污染的物种来说组织内和血液内是相同的。作者发现大肠杆菌在人类微生物群系中普遍存在,在组织(64.68%)和血液(66.29%)中大肠杆菌序列的比例几乎相同,且与测序中心密切相关,作者还发现组织和血液中大肠杆菌序列的读数率是一致的并且覆盖度也类似。不过,少数大肠杆菌基因显示出类似于组织定植微生物群的特征,同时在血液中大肠杆菌基因分布呈双峰性,表明大肠杆菌存在组织定植和污染两种分布情况。
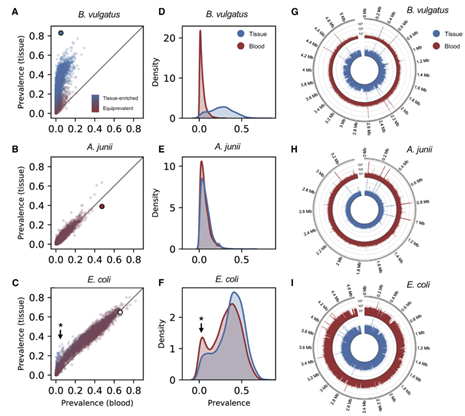
7、全基因组测序(WGS)和全外显子测序(WXS)显示CRC携带的大肠细菌与血液和大脑不同
CRC组织和血液样本内的突变的分布很大程度上重现了物种和基因水平的结果,而且作者还在大肠杆菌基因组中发现了明显的组织特异富集和组织与血液相当的变异,这表明在混合证据的情况下,序列变异分析可能证明在内源和同源基因测序之间没有区别。
8、序列去污染去除了来自测序中心的差异
作者在去污染后发现去除污染对所有样品均有影响,但细菌丰度较低的样品受影响最大,去污染还规范了CRC组织样本的相对丰度分布,最显著的是去除污染物放线菌和变形菌。作者从原始TCGA测序数据中提取的组织留置成分经非监督聚类显示其分布不依赖于测序中心而与样本类型相关,同时检查污染成分时,作者发现样本不再按样本类型聚类,而是完全按照测序中心进行分布。作者发现其算法极大地增加了来自不同测序中心的患者的匹配的组织样本序列中微生物种群之间的相似性,同时保持了样本之间的总体多样性,证明了算法的可靠性。
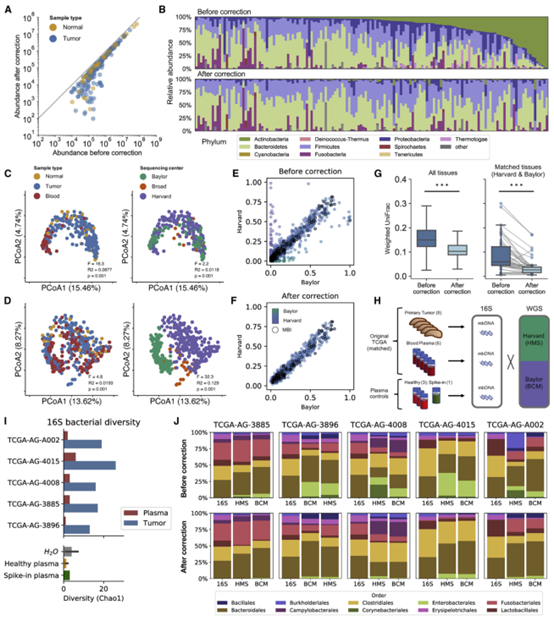
作者应用原始TCGA组织和血液样本验证了组织中富集的微生物成分,同时验证组织和血液同时分布的物种是污染物。使用Sparcc 作者发现梭杆菌(Fusobacterium)和拟杆菌(Bacteroides)在结直肠组织微生物群中特异性相关。
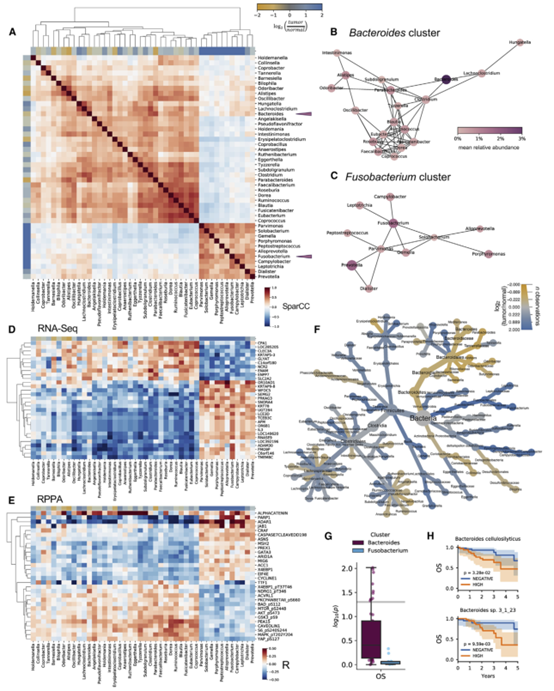
作者使用来自TCGA的同一批宿主的归一化RPPA、mRNA-seq、miRNA-seq和甲基化27-K数据来探索这些数据指标与之前鉴定的梭杆菌和拟杆菌群属之间的相关性,发现细菌互作与宿主基因表达模式有相关性。Fusobacterium 互作菌群和Bacteroides互作菌群可能代表了不同的免疫调控和病原反应。作者通过匹配的肿瘤-正常分组比较发现了F. nucleatum,F. hwasookii,一些未分类的Fusobacterium菌,还有P. micra, S. moorei,和 P. stomatis 等都与结直肠肿瘤相关。同时,拟杆菌互作组的物种通常比梭杆菌互作组的菌对宿主有更好的预后性。
9、CRC组织中的微生物与宿主免疫反应、炎症性癌变途径和细胞-细胞粘附有关
作者为了论证之前发现的显著富集在癌症组织或者正常组织的菌(37种)是否对机体基因的表达模式或者通路有一致的影响。作者在159个CRC肿瘤样本中将这些物种的丰度与匹配样本中mRNA表达数据做了相关性分析,发现了与肿瘤和正常组织相关物种相关的转录模式,基于此转录模式的差异,作者又通过GSEA分析识别了二者相关的表达通路。通路分析作者发现(1)与细菌共相关的基因一致地富集在免疫系统激活地通路和过程中,而不管他们与肿瘤和正常组织的关系;(2)肿瘤相关基因和正常组织相关基因分别富集在与炎症性癌症通路和细胞粘附相关过程的通路里;(3)肿瘤和正常组织相关物种都富集了与肠道IgA生成、抗原递呈、自然杀伤细胞介导的细胞毒性、细胞因子信号转导和原发性免疫缺陷相关的过程,表明这些细菌的存在几乎普遍激活了免疫原性转录反应;(4)作者还发现,包括DNA复制、DNA修复、氧化磷酸化、p53信号通路和核糖体活性在内的通路在正常组织相关物种中均呈负富集,而在肿瘤相关物种中呈正富集,与梭杆菌有关的基因尤其如此。
作者发现CRC患者血液样本中的细菌明显比BC患者更丰富和多样,作者推测粘膜屏障功能缺失是CRC和其他慢性炎症疾病的共同特征,可能导致微生物从CRC转移到固有层和血流。为验证这个推测,作者下载了已报道的共生菌(407个)和黏膜损伤相关菌(693个)并检测两个菌集在CRC病人和BC病人样本的分布。作者发现在CRC 血液样本中,黏膜损伤相关的菌更普遍。作者又比较粘膜损伤相关的菌与共生菌的丰度,发现黏膜损伤的菌在CRC血液样本中比BC血液样本更普遍的高丰度。
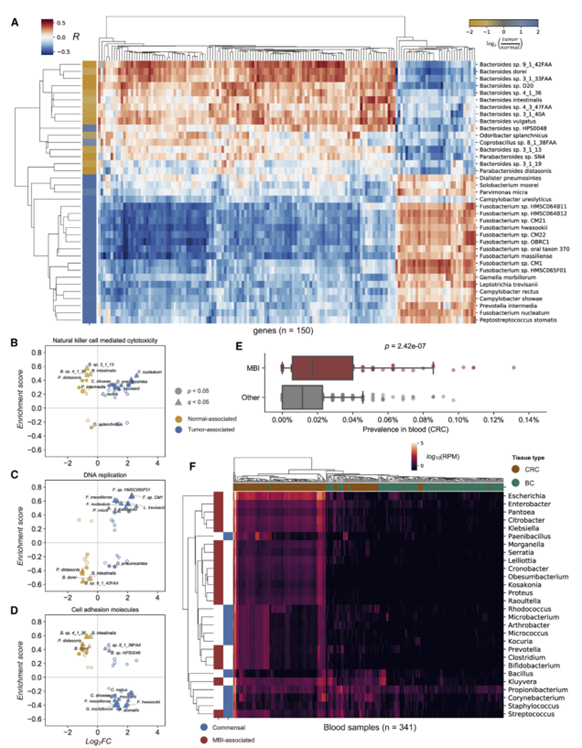
10、污染调整后的TCGA消化道癌组织微生物组特征
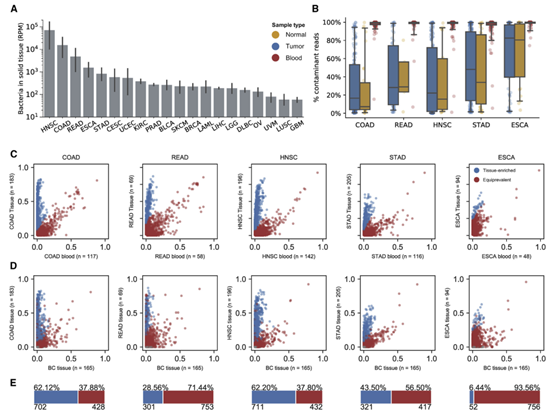
成功识别CRC组织定植的菌后作者看了TCGA里面所有肿瘤类型的肿瘤组织里面的菌,结果发现胃肠道癌症组织定植的菌是最多的,尤其头颈癌(HNSC)、结肠癌(COAD)、直肠癌(READ)、食道癌(ESCA)和胃癌(STAD)的组织样本在去污染前有最多的细菌序列,而葡萄膜黑色素瘤(UVM)、肺鳞状细胞瘤(LUSC)和胶质母细胞瘤最少。作者进一步去掉胃肠道癌症组织的污染序列,经与血液样本和脑癌组织比较发现了组织特异性定植的物种(如下图)。在膀胱癌(BLCA)、乳腺癌(BRCA)、子宫癌(UCEC)、子宫癌(CESC)或前列腺癌(PRAD)中,很少能检测到具有统计学意义的组织定植菌(如下图),由于这些组织菌量本来就很低,因此在这些组织中分离出污染序列也很有挑战性。
总结
作者通过比较和整合来自多个NGS平台和不同样本类型的数据,分离并通过实验验证了这些数据集的组织定植成分,产生了一个净化了的TCGA组织样本微生物公共资源数据库TCMA。TCMA是一种前所未有的资源,为研究者探索组织定植菌群在各种癌症类型中的作用,并识别可预测的微生物生物标志物提供了可靠的支持。癌症组织中微生物可用于预测宿主组织分子环境、临床结果、宿主免疫效应、炎症性癌症通路、细胞-细胞黏附、黏膜屏障损伤等,大家后续可以应用起来了。
科研培训